
Abstract
Autoimmune diseases are immune disorders characterized by T cell hyperactivity and B cell overstimulation leading to overproduction of autoantibodies. Although the pathogenesis of various autoimmune diseases remains to be elucidated, environmental factors have been thought to contribute to the initiation and maintenance of auto-respond inflammation. Toll-like receptors (TLRs) are pattern recognition receptors belonging to innate immunity that recognize and defend invading microorganisms. Besides these exogenous pathogen-associated molecular patterns, TLRs can also bind with damage-associated molecular patterns produced under strike or by tissue damage or cells apoptosis. It is believed that TLRs build a bridge between innate immunity and autoimmunity. There are five adaptors to TLRs including MyD88, TRIF, TIRAP/MAL, TRAM, and SARM. Upon activation, TLRs recruit specific adaptors to initiate the downstream signaling pathways leading to the production of inflammatory cytokines and chemokines. Under certain circumstances, ligation of TLRs drives to aberrant activation and unrestricted inflammatory responses, thereby contributing to the perpetuation of inflammation in autoimmune diseases. In the past, most studies focused on the intracellular TLRs, such as TLR3, TLR7, and TLR9, but recent studies reveal that cell surface TLRs, especially TLR2 and TLR4, also play an essential role in the development of autoimmune diseases and afford multiple therapeutic targets. In this review, we summarized the biological characteristics, signaling mechanisms of TLR2/4, the negative regulators of TLR2/4 pathway, and the pivotal function of TLR2/4 in the pathogenesis of autoimmune diseases including rheumatoid arthritis, systemic lupus erythematosus, systemic sclerosis, Sjogren’s syndrome, psoriasis, multiple sclerosis, and autoimmune diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
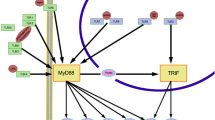
Toll-like receptors are membrane-bound proteins that recognize invading organisms bearing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [1]. PAMPs are conserved molecules derived from microorganisms, for example, lipopolysaccharide (LPS), peptidoglycan, flagellin, and microbial nucleic acids, while most DAMPs are endogenous molecules released from dying host cells molecules upon cellular stress or tissue damage, such as oxidative stress and heat shock proteins [2]. Activation of Toll-like receptors (TLRs) by PAMPs or DAMPs can up-regulate inflammatory cytokines and chemokines, and engage an assortment of intracellular signaling pathways to regulate the nature, magnitude, or duration of the host’s inflammatory response [3]. The first TLR to be characterized was Toll-like receptor 4 (TLR4) by Medzhitov in 1997 [4]. Thereafter, 13 TLRs have been identified, TLR1 to TLR13, among which TLR1 to TLR10 functions in human [5]. TLRs play an essential role in innate immune system. They regulate a pro-/anti-inflammatory balance [6] (Fig. 1).

TLR2/4 signaling pathway and negative regulators [17–43]. PAMPs or DAMPs bind with TLR2-TLR1/TLR2-TLR6 heterodimer or TLR4 homodimer to activate MyD88-dependent signaling pathway that recruits TIRAP/MyD88 complex and its downstream kinases, the IRAK1/IRAK4 complex. After that, TRAF6 is recruited to activate IKKs complex and TAK1/TAB complex, which lead to the nuclear translocation of NF-κB and activation of MAPKs. Ligation of TLR4 homodimer can also activate MyD88-independent signaling pathway in which TRAM is required for the activation of TRIF which interacts with TRAF3 and TRAF6. Activation of TRAF6 and RIP1 leads to the nuclear translocation of NF-κB, while TRAF3 induces activation of TBK1/IKKi with subsequent nuclear translocation of IRF3. In this signal transduction process, endogenous inhibitors negatively regulate TLR2/4 signaling. SOCS1 facilitates ubiquitination and proteasomal degradation of TIRAP. MyD88s cannot associate with IRAK4 and prevents IRAK4 from phosphorylating IRAK1. ADAM15 acts as a negative regulator of TRIF. Tollip reduces the autophosphorylation of IRAK-1. IRAK-M lacks the kinase activity of its counterparts IRAK1 and IRAK4, thus silences signal transduction. SHP-1 causes inactivation of IRAK1 through binding to its kinase tyrosine-based inhibitory motif (KTIM). CD300a and CD300f can function through SHP-1 to inhibit IRAK activity. A20 deubiquitinates TRAF6. USP25 reverses the Lys(48)-linked ubiquitination of TRAF3. SyK diminishes TRAF6-dependent proinflammatory signaling while elevated TRAF3-dependent IFN production. Triad3A promotes substantial degradation of TLR4, decreases TRAF3 level and blocks IRF-3 activation
Autoimmune disease is an inflammatory disorder that characterized by the production of autoantibodies. Although the mechanism of autoimmune diseases remains elusive, accumulated evidence implicates the association between TLRs and autoimmune [7]. TLR2 and TLR4 are cell-surface TLRs. Recent studies have demonstrated them to be significant in the pathogenesis of autoimmune diseases. In this review, we summarized the biological characteristics, signaling pathway, and inhibitors of TLR2/4 and their function in autoimmune diseases (Tables 1 and 2).
Structure of TLRs
All TLRs are integral membrane glycoprotein receptors with molecular weights ranging from 90 to 150 kDa. They are composed of three components: an N-terminal ligand recognition domain, a single transmembrane helix, and a C-terminal cytoplasmic signaling domain [8]. The recognition domain contains 16–28 leucine-rich repeats which form a horseshoe shape and is responsible for the recognition of PAMPs and DAMPs [9]. The transmembrane domain of TLRs contains atypical stretch of approximately 20 uncharged, mostly hydrophobic residues [10]. The signaling domain of TLRs is also known as Toll/interleukin-1 receptor (TIR) domain because they are homologous with interleukin-1 receptors (IL-1R) [11]. TLR2 and 4 are both cell-surface TLRs with extracellular recognition domain. Engagement of ligands with the TLR2/TLR1 or TLR2/TLR6 heterodimer or the TLR4 homodimer induces activation of intracellular “signaling cascades” when the TIR domain is attached by intracellular adaptors [12].
Ligands of TLR2/TLR4 and Their Signaling Pathways
TLR2 and 4 are expressed by a wide range of cell types including professional immune cells, for example, monocytes, myeloid DCs, mast cells, as well as T and B lymphocytes, and nonprofessional immune cells like synovial fibroblast-like cells and epithelial cells [13]. As an important member of pattern recognition receptors, TLR2 can bind a wide range of both exogenous ligands including lipoproteins, peptidoglycan, lipoteichoic acid, lipoarabinomannan, glycosylphosphatidylinositol, phenol-soluble modulin, zymosan, glycolipids, and endogenous DAMPs, such as Snapin, Hyaluronic acid, heat shock protein (HSP)70, and high mobility group box protein 1 (HMGB1) [13, 14]. However, TLR4 can recognize and bind different ligands, for instance, the exogenous PAMPs like lipopolysaccharide, taxol, viral glycoproteins, rSV fusion protein, mice mammary tumor virus (MMTV) envelope protein, as well as endogenous ligands, such as necrotic cells, heat-shock proteins, HMGB1, fibronectin, extracellular cell matrix (ECM) components, fatty acid, minimally oxidized low-density lipoprotein (mmLDL), and fibrinogen [15, 16].
The ligation of TLRs can recruit five cytosolic adaptors: myeloid differentiation primary response 88 (MyD88), TIR domain-containing adaptor-inducing IFN-\( \beta \)(TRIF), TIR domain-containing adaptor protein (TIRAP; also known as MAL), TRIF-related adaptor molecule (TRAM), and Sterile \( \alpha \) and armadillo motif-containing protein (SARM) [17]. The canonical TIR pathway depends on MyD88, which is utilized by all TLRs, except for TLR3. Upon activation in MyD88-dependent pathway, both TIRAP/MAL and MyD88 are recruited through TIR–TIR interactions to the TLR2/TLR1 or TLR2/TLR6 heterodimer or the TLR4 homodimer [18]. MyD88 interacts with interleukin-1 receptor-associated kinase (IRAK) complex, which consists of two active kinases (IRAK-1 and IRAK-4) and two noncatalytic subunits (IRAK-2 and IRAK-3/M) [19]. The phosphorylation of IRAK-4 can in turn activate IRAK-1 and then recruit tumor necrosis factor receptor-associated factor 6 (TRAF-6) to form MyD88–IRAKs–TRAF-6 complex which activates a complex containing TGF-β-activated kinase 1 (TAK1), TAK1-binding protein 1 (TAB1), TAB2, and TAB3 [3, 20]. Activation of the TAK1/TAB complex activates both the mitogen-activated protein kinases (MAPKs) and the inhibitor of NF-κB kinase (IKK) complex [21]. IKK complex is made up of IKKα, IKKβ, and IKKγ/NEMO (NF-κB essential modulator) [22]. Activation of IKK complex leads to the phosphorylation of the inhibitor of NF-κB (IκB) proteins, which results in the ubiquitination and subsequent degradation of IκB [23]. Thus, the NF-κB gets free and translocates into nucleus to regulate gene transcription and inflammatory cytokines and chemokines production, such as IL-1β, IL-6, IL-8, IL-12, IL-17, TNF-α, IFN-γ, iNOS, and ICAM-1 [24]. However, a research group found that activation of NF-κB by TLR4 signaling is specifically TAK1 independent in synovial fibroblasts in rheumatoid arthritis [25].
In MyD88-independent signaling pathway, TLR4 needs TRAM for the activation of TRIF that interacts with TRAF3 and TRAF6 [26]. The activation of TRAF6 and receptor-interacting protein1 (RIP1) leads to the nuclear translocation of NF-κB [27]. The interaction between TRAF3 and TBK1/IKKi causes the phosphorylation of IRF3 leading to its translocation to nucleus and initiate the production of type IFN [28]. In this process, SARM acts as a negative regulator of TRIF activation [29]. Additionally, TRAF3 differently controls MyD88- and TRIF-signaling; the ubiquitination and degradation of TRAF3 have been demonstrated to be essential in MyD88-dependent pathway [30].
Endogenous Negative Regulators of TLR2 and TLR4 Signaling
However, there are some endogenous inhibitors that modulate TLRs signaling pathways, which are important for the maintenance of the balance of immune response. These inhibitory proteins target the receptors, adapter molecules, and key kinases to impair the TLR-mediated production of inflammatory cytokines.
Regulators Target Adaptors
The most universal adaptor molecule in TLR signaling is MyD88. After being stimulated by LPS or TNF, a splice-variant of MyD88, MyD88s, is up-regulated in monocytes. This short form of MyD88 cannot associate with IRAK4 and prevents IRAK4 from phosphorylating IRAK1, thereby preventing the activation of NFκB [31]. And the partner of MyD88, adaptor TIRAP, can be inhibited by suppressor of cytokine signaling (SOCS) 1. It facilitates ubiquitination and proteasomal degradation of TIRAP. In vivo experiment demonstrates that cells from SOCS1 transgenic mice are unable to respond to LPS [32]. In MyD88-independent pathway, adaptor protein TRIF can be inhibited by A disintegrin and metalloprotease (ADAM)15 which acts as a negative regulator of TRIF-mediated NF-κB and IFN-β reporter gene activity possibly by mediating the proteolytic cleavage of TRIF. Suppression of ADAM15 expression can enhance TRIF-mediated cytokine production [33].
Regulators Target IRAKs
Downstream of these adaptors, tyrosine phosphatase SHP-1 causes inactivation of IRAK1 through binding to its kinase tyrosine-based inhibitory motif (KTIM) [34]. Additionally, CD300a and CD300f, membrane of CD300 family, can function through SHP-1 to inhibit MyD88-mediated signaling. However, CD300f can also activate SHP-2 to regulate TRIF-mediated TLR signaling pathways which required the combined action of SHP-1 and SHP-2 [35]. A substrate of SHP-1, SHPS-1, can also negatively regulate the MyD88-dependent TLR signaling pathway. The inhibitory effects were modulated by SHPs and phosphoinositide 3-kinase (PI3K) [36]. Another two inhibitors targeting IRAKs are IRAK-M and Toll-interacting protein (Tollip). IRAK-M belongs to IRAK kinase family but lacks the kinase activity of its counterparts IRAK1 and IRAK4. It exhibits suppressive capacity to the production of proinflammatory cytokines and inhibits downstream NFκB activation [37]. Tollip targets IRAK1 and reduces the autophosphorylation of IRAK-1. Moreover, some studies showed that Tollip was also able to interact with TLR2 and TLR4 [38].
Regulators Target TRAFs
A20 (also referred to as TNF-α-induced protein 3; TNFAIP3) is an ubiquitin-editing enzyme that specifically deubiquitinates TRAF6, thereby inhibiting its downstream TLR signaling [39]. And regulator ubiquitin-specific protease 25 (USP25) has been identified to inhibit TRAF3. USP25 is recruited to the TLR4 signaling complex upon the ligation of TLR4 and LPS. And it down-regulates TLR4 signaling by reversing the Lys(48)-linked ubiquitination of TRAF3 [40]. Non-receptor tyrosine kinase Sky also targets TRAFs and differently regulates the LPS-dependent, lysine 63-linked ubiquitination of TRAF6 and TRAF3, which results in the diminished TRAF6-dependent proinflammatory signaling and elevated TRAF3-dependent IFN production [41].
Regulator Targets Multiple Molecules
Triad3A is an E3 ubiquitin ligase that interacts with several components in TLR signaling pathway. Triad3A can be induced following decreased TRAF3 level in a dose-dependent manner. In one hand, Triad3A overexpression promotes substantial degradation of TLR4 with a concomitant decrease in signaling, but does not affect TLR2 expression or signaling [42]. In the other hand, Lys48-linked ubiquitination of TRAF3 by Triad3A increases TRAF3 turnover, and Triad3A expression blocks IRF-3 activation by Ser-396 phosphorylation [43].
All those negative regulators of TLR signaling pathway restrict immune responses and enable a balanced immune homeostasis.
TLR2 and TLR4 in Autoimmune Diseases
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a progressive inflammatory autoimmune disease mainly affecting the joints. It is characterized by synovial hyperplasia and inflammatory cell infiltration, leading to tissue destruction and functional disability [44]. Environmental factors have been proposed to be triggers of inflammatory response in RA and contribute to the persistence of inflammation [45]. T cells, B cells, macrophages, and fibroblast-like synoviocytes (FLSs) are important mediators of chronic inflammation of RA [46]. TLR2 and TLR4 have been demonstrated to be expressed on macrophages and synovial fibro-blasts and the expressional levels increase in RA patients than osteoarthritis (OA) patients and healthy donors [47, 48]. And the TLR4 responsiveness to LPS stimulation was also enhanced in peripheral blood mononuclear cells from patients with recent onset rheumatoid arthritis than in cells from OA and healthy control subjects, which caused the dramatically increase of IL-6 and TNF-alpha production [49]. TLR2 activation promotes angiogenesis, cell adhesion, and invasion through the Tie2 signaling pathway, key mechanisms involved in the pathogenesis of RA [50].
Exogenous ligands like peptidoglycan, lipoteichoic acid, and lipopolysaccharides binding with TLR2 or TLR4 can enhance the production of pro-inflammatory cytokines and chemokines such as interleukin 6 (IL-6) and interleukin 17 (IL-17) in human synovial fibroblasts and peripheral blood mononuclear cells (PBMC) from RA patients [51, 52] and trigger cartilage inflammation and degeneration [53]. This may explain how the microbial trigger involved in the pathogenesis of RA. In addition to the potential role of microbial TLR ligands, a number of endogenous proteins have been implicated as potential endogenous TLR ligands in RA. Cells under strike during synovial inflammation can release production of heat shock proteins (HSPs), high mobility group box protein 1 (HMGB1), hyaluronan fragments, and hypoxia-inducible factor-1α (HIF-1α) [54, 55]. Hsp70 and Hsp72 released from FLSs in response to heat shock or TNF-α may be a major paracrine/autocrine inducer of IL-10 production in FLSs via TLR2 or TLR4 [56]. Heat shock glycoprotein 96 (Gp96) expression increased in synovial fluid from the joints of RA compared with disease controls and activated macrophages through TLR2 signaling, which strongly correlated with inflammation and synovial lining thickness [57]. HMGB1 is a non-histone nuclear protein that can serve as an alarm to drive the pathogenesis of inflammatory and autoimmune diseases. HMGB-1 expressional level increased in RA and its ligation with TLR4 enhanced production of TNF, IL-6, IL-8, and matrix metalloproteinase (MMP) 3 in RASFs [58]. Additionally, HMGB-1 can promote the differentiation of Th17 via up-regulating TLR2 and IL-23 of CD14+ monocytes from patients with RA [59]. Hyaluronic acid (HA) differently modulates TLR-4 and the inflammatory response depending on its degree of polymerization. Small HA fragments initiate pro-inflammatory responses, while highly polymerized HA exert a protective effect in inflammatory pathologies such as rheumatoid arthritis [60]. Thus, the release of endogenous TLR ligands creates a vicious circle of inflammation.
Animal models also afford evidence for the importance of TLR2 and TLR4 in RA development. Different TLR knockouts in the IL-1 receptor antagonist knockout model show opposite effects: IL1rn−/−Tlr2−/− mice developed a more severe arthritis with reduced inhibitory function of regulatory T cells (Tregs) and dramatically increased IFN-gamma production by T cells, while IL1rn−/−Tlr4−/− mice were protected against severe arthritis with markedly lower numbers of Th17 cells and a impaired capacity to produce IL-17 [61]. In collagen-induced arthritis mice model, TLR4 activation by LPS enhanced the protease high temperature requirement A1 (HTRA1) expression through the NF-κB pathway in fibroblasts and macrophages and increased the incidence of collagen-induced arthritis in mice [62]. And the incidence and severity of collagen- induced arthritis was significantly lower in TLR4-deficient DBA1J mice compared to wild-type mice [63]. These results imply that blockade of TLR4 and induction rather than blockade of TLR2 may be new therapeutic strategies.
Recent years, numerous research groups have joined forces in an endeavor to discover the correlation between polymorphisms of TLRs and RA susceptibility or severity. Most of these studies focused on two functional variants of TLR4: Asp299Gly and Thr399Ile (D299G/T399I) [64]. However, the results are not in consistence [65, 66]. So, in order to determine whether toll-like receptor polymorphisms confer susceptibility to RA and influence the clinical characteristics of RA, some research groups performed meta-analysis. The results show lack of an association between the TLR4 polymorphism and RA but the numbers of guanine-thymine [(GT)(n)] repeats in intron II of the TLR2 gene presented a significantly higher S-allele frequency in Korean patients with RA than in controls (30.3 vs. 23.0 %, p = 0.03) [67, 68]. Besides this, another study identified two non-missense genetic polymorphisms located in regulatory region of TLR4 (minor allele C and homozygotic variant genotype CC of rs41426344 and minor allele C of rs7873784) to be risk factors for the development of RA in Chinese Han people, which suggests the non-missense polymorphisms located in regulatory region should not be ignored in disease association analysis [69].
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a non-organ-specific autoimmune disease characterized by B cell hyperactivity, abnormally activated T cells, defects in the clearance of apoptotic cells, and immune complexes [70]. SLE is thought to result from interaction between genetic and environmental factors [71]. SLE has an increased overall risk for infection and bacterial infections in SLE are associated with higher morbidity and mortality [72]. One of the mechanisms whereby bacteria and viruses can participate in autoimmune disorders is their interaction with TLRs. Recently, some studies revealed the expressional change of TLR2/4 in SLE. Komatsuda A et al. found that the mRNA level of TLR2 significantly increased in PBMCs of SLE patients, while Martina Kirchner et al. identified that the surface expression of TLR4 on CD14+ monocytes decreased dramatically in SLE patients compared with control subjects [73, 74]. Moreover, the number of CD180-negative B cells (CD180 molecule is a homologue and negative regulator of TLR4) in peripheral blood changes in parallel with disease activity in SLE patients and the animal experiment testified that CD180-negative B cells contributed significantly to anti-dsDNA and histone antibodies production and renal lesions [75].
Cytokines and autoantibodies production from PBMCs in response to TLR activation is dysregulated in SLE patients. It was found that in SLE patients IL-10 protein production was down-regulated after the activation of TLR-2 and TLR-4, but TNF-α protein production was decreased after the activation of TLR-2 rather than TLR-4 [76]. And the endogenous ligand HMGB1 in circulating DNA-containing immune complexes (ICs) is crucial for anti-dsDNA antibody induction depending on the TLR2/MyD88/miR-155/Ets-1 pathway [77]. In addition, Loser K et al. found that local myeloid-related protein-8 (Mrp8) and Mrp14 can induce autoreactive CD8+ T cells and increase interleukin-17 (IL-17) expression via Toll-like receptor 4 signaling, which is essential for the development of systemic autoimmunity [78].
Experimental evidence in animal models for SLE suggests a role for TLR2 and TLR4 in the development of murine lupus. Compared to C57BL/6lpr/lpr mice, TLR2- and TLR4-deficient C57BL/6lpr/lpr mice develop a less severe disease, fewer immunological alterations, a diminished renal lesion, and significantly reduced antinuclear, anti-dsDNA, and anti-cardiolipin autoantibody titers [78]. Particularly in C57BL/6lpr/lpr-TLR4-deficient mice, analysis of B cell phenotype showed a significant reduction of marginal zone B cells and a decrease of IL-6 production [79]. Conversely, activation of TLR2/4 by proteoglycan biglycanin MRL/lpr mice can trigger CXCL13 expression and cause accumulation of B cells with an enhanced B1/B cell ratio in the kidney [80]. In transgenic mice with monoclonal anti-dsDNA, ligation of TLR4 by lipopolysaccharides in vitro and in vivo induced severe SLE syndromes through the overproduction of IL-10 and IFN-gamma [81]. Compared to wild-type, TLR4−/− mice injected with pristine (an experimental model used to study lupus nephritis) demonstrated a global decrease in Th1, IFN-gamma, and IL-17A and IL-6 cytokine production and autoantibody levels of anti-dsDNA and anti-RNP were both down-regulated andrenal injury was attenuated in TLR4−/− mice which showed less glomerular immunoglobulin and complement deposition. The results indicated that TLR4 was required for the development of autoimmunity and lupus nephritis in pristane nephropathy [82].
Although the polymorphisms of TLR2 (Arg677Trp and Arg753Gln) and TLR4 (Asp299Gly and Thr399Ile) genes lack association with the susceptibility or severity of SLE, the TIRAP single nucleotide polymorphism rs8177374 (C/T), which encodes a leucine substitution at serine 180 of Mal (S180L), is demonstrated to be a protective factor against developing systemic lupus erythematosus [83].
Systemic Sclerosis
Systemic sclerosis (SSc) is a connective tissue disease of presumed autoimmune origin characterized by excessive extracellular matrix deposition in the skin and other visceral organs [84]. Fibrosis is the central process of scleroderma or systemic sclerosis resulting from inflammatory cell infiltration and the release of cytokines and growth factors causing fibroblasts activation and enhanced extracellular matrix (ECM) synthesis and deposition [85]. Recent studies implicated that activation of Toll-like receptors contributed to the pathogenesis of fibrosis. Activation of TLR4 enhanced IL-10 secretion by TLR4-stimulated DCs, which led to elevated CCL18 levels in SSc patients [86]. Also, stimulation of DC subsets from patients with early SSc by ligands for TLR2 or TLR4 resulted in increased secretion of IL-6, IL-10 as well as TNFalpha and a decreased IL-12 production [87]. Besides DCs, TLR4 is also expressed on surface of fibroblasts and activation of TLR4 contributes to the up-regulation of production of profibrotic and proangiogenic chemokines by fibroblasts [88]. Bhattacharyya S et al. found that activation of fibroblast TLR4 signaling with damage-associated endogenous ligands could augment fibroblast sensitivity to transforming growth factor-b1 (TGF-b1) and promote matrix production and connective tissue remodeling by increasing fibrogenic genes expression and inhibiting anti-fibrotic microRNA expression which reveals the role of TLR4 in converting self-limited tissue repair into intractable fibrosis [89]. Besides endogenous ligation of TLR4, environmental stimuli omniscan and gadodiamide signaling via TLR4 and 7 could enhance production of numerous proinflammatory or profibrotic cytokines, chemokines, and growth factors, including CXCL10, CCL2, CCL8, CXCL12, IL-4, IL-6, TGF-b, and vascular endothelial growth factor [90]. In animal model of human systemic sclerosis, skin fibrosis induced by subcutaneously injected with bleomycin in vivo, is attenuated in mice harboring a missense mutation in the intracellular signaling domain of TLR4 [89]. And CD19 deficiency suppresses fibrosis and autoantibody production in mouse model by inhibiting TLR4 signals [91]. Therefore, TLR4 may be a potential therapeutic target for SSc.
Sjogren’s Syndrome
Sjogren’s syndrome (SjS) is a chronic slowly progressing autoimmune disease, characterized by dysfunction and destruction of salivary and lacrimal glands associated with chronic lymphocytic infiltrating lesions, resulting in dry eyes and dry mouth [92]. In addition to the apparent primary sites of autoimmunity in SjS, other organs can be involved in the pathologies of SjS including the lungs, kidneys, GI tract, skin, vasculature, bladder, and vagina [93]. In salivary gland epithelial cells (SGEC) and acinar cells as well as salivary-infiltrating mononuclear cells of SjS patients, TLR2 and TLR4 display significant higher expressional levels than controls [94–96]. In cultured human salivary cells (HSG), a similar expression pattern is observed. Agonists to TLR stimulate CD54 expression and IL-6 production through phosphorylation of MAPKs in HSG cells rather than Akt phosphorylation or activation of NF-kappaB p65 [94]. In addition, stimulation of TLR2 induces the production of IL-23/IL-17 from the PBMCs of primary SjS patients via IL-6, STAT3, and NF-kB pathway [95]. All these data indicate that TLR2 and TLR4 contribute to the pathogenesis of Sjogren’s syndrome.
Psoriasis
Psoriasis is an immune-mediated skin disease characterized by abnormal keratinocyte differentiation and proliferation [97]. The roles of TLR2 and TLR4 in psoriasis are implied by the elevated expression of TLR2 and TLR4 on peripheral blood mononuclear cells and keratinocytes in patients with psoriasis [98, 99].
Multiple Sclerosis
Multiple sclerosis (MS) is considered a chronic inflammatory disease of the central nervous system with autoimmune origin; it is characterized by inflammation, demyelination, and axonal pathology [100]. Bacterial and viral infections are potential cofactors implicated in the initiation and persistence of autoimmune inflammation [101]. Accordingly, TLRs, an important member of pattern recognition receptors family, are hypothesized to be involved in the pathogenesis of MS. One of the evidence supports this hypothesis is the elevated expressional level of TLR2 and TLR4 in MS patients and rodent experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis [101, 102]. Moreover, TLR2 is required for repressive effects of hyaluronan on oligodendrocyte precursor cells maturation in vitro and TLR2-null mice showed enhanced remyelination in the lysolecithin remyelination model [102]. Another TLR2/4 ligand, HMGB-1, is evident in active lesions of MS and EAE and functions as a potent proinflammatory signal through interaction with TLR2 or TLR4 [103]. However, in EAE model, the loss of TLR4 solely in CD4(+) T cells almost completely released disease symptoms or even could not induce EAE when transferred into Rag1−/− mice, mainly through diminished Th17 and, to a lesser degree, Th1 responses. Moreover, compared with WT γδ T cells, Tlr4(−/−) γδ T cells exhibited almost 50 % reduction in IL-17 and IFN-γ production following EAE induction [104]. TLR4 signaling pathway also plays a role in the response to interferon-beta (IFNβ) treatment in MS patients. Baseline expression of the interleukin-1 receptor-associated kinase 3 (IRAK3), a negative regulator of TLR4 signaling, has been demonstrated to be significantly decreased in IFNβ responders compared with no responders [105]. Thus, inappropriate responses of TLR2 or TLR4 have been testified to be involved in the pathogenesis of multiple sclerosis.
Autoimmune Diabetes
Type 1 diabetes (T1D) is characterized by autoimmune destruction of the insulin-secreting beta cells in the pancreas [106]. Recent studies demonstrated that TLR2/4 could serve as a link between innate immunity and pro-inflammatory state of T1D [107]. The mRNA expressional levels of TLR2, TLR4, and MyD88 as well as ligands of TLR2 or TLR4, such as Hsp60 and HMGB1, have been identified elevated in patients with T1D [108, 109]. Also, TLR4 expressed mainly on β cells so that ligands, like HMGB1, may signal via TLR4 to selectively damage β cells rather than α cells during the development of T1D, which contributes to the initiation of T1D [110]. Compared to non-diabetic (ND) donors, B cells from diabetic patients produce a higher level of IL-8 and a lower level of IL-10 in response to TLR2 and TLR4 activation [111]. TLR4 is also associated with lipid metabolism [112]. TLR4 deficiency reduced lipid accumulation in cardiac muscle early after the onset of type 1 diabetes, with a decreased level of lipoprotein lipase (LPL) and an increase level of phosphorylation of AMP-activated protein kinase (AMPK) [113]. Evidence from mice experiments also indicates the important role of TLR2 and TLR4 in the pro-inflammatory state of T1D. Knockout of TLR2 or TLR4 in mice attenuates the production of pro-inflammatory cytokines in streptozotocin-induced diabetic mouse model [114, 115]. Additionally, some researchers found that late apoptotic β-cells with secondary necrosis could induce inflammatory responses in macrophages through the toll-like receptor 2/MyD88/nuclear factor-κB signaling pathway. It could also promote TLR2-dependent maturation of dendritic cells, and then activate autoreactive T-cells. However, in TLR2 knockout mice apoptotic β-cells failed to prime diabetogenic T-cells in the pancreatic lymph nodes, thereby conferring a significant protection against type 1 diabetes and insulitis in T1D animal models [116]. To reduce TLR2 responsiveness and induce TLR tolerance, some research groups treated NOD mice with TLR2 agonists, Pam3CSK4 [117, 118]. Chronic Pam3CSK4 administration successfully prevented the development or recurrence after islet transplantation of diabetes in NOD mice and inhibited priming of naive T cells as well as the activity of sensitized T cells [118]. Moreover, treatment of prediabetic mice with a synthetic TLR2 agonist enhanced the number and function of CD4+CD25+ Tregs and conferred DCs with tolerogenic properties by which diminished T1D. TLR2 ligation also promoted the expansion of Tregs upon cultured with DCs and enhanced their capacity to prevent the disease [117]. All these data demonstrate the central role of TLR2 and TLR4 in the pathogenesis of autoimmune diabetes and indicate new therapeutic perspectives.
Therapeutic Perspectives
A bulk of studies suggests that TLR2 and TLR4 pathways contribute to the development of autoimmune diseases. The underlying mechanisms are becoming clearer and they offer exciting therapeutic options. TLR2 and TLR4 as well as their ligands, adaptors and downstream kinases, negative regulators, and even microRNAs targeting TLRs could be intervention targets. Small molecules activate or inhibit these targets have been identified and summarized in previous review [119]. There are also some traditional Chinese medicine extractives which can inhibit inflammation through TLR pathway. Celastrol, a pentacyclic-triterpene extract from Tripterygium wilfordii Hook might inhibit FLS migration and invasion by suppressing TLR4/NF-kB-mediated MMP-9 expression [120]. And coumarins, the major components of Urtica dentate Hand, can maintain the DCs in an immature tolerogenic state partially by down-regulating TLR4-signaling pathways in DCs and promote Treg differentiation [121]. Different molecules can target different components in TLR2 or TLR4 signaling pathways to regulate inflammatory responses, thereby ameliorating autoimmune diseases.
Conclusion
Increasing evidence about TLR2 and TLR4 has led to the recognition that the innate immune system may act, under some circumstances, as a double-edged sword. In addition to its beneficial role in host defense, it may lead to initiation and maintenance of autoimmune responses. Treatments target TLR2 and TLR4 signaling can break the perpetuated inflammatory loop and ameliorate autoimmune diseases. But exact mechanism underlying the interaction between TLR2 and TLR4 with autoimmune diseases remains unclear; therefore continued efforts in this direction are required.
References
Sabroe I, Read RC, Whyte MK, Dockrell DH, Vogel SN, Dower SK (2003) Toll-like receptors in health and disease: complex questions remain. Journal of immunology (Baltimore, Md : 1950) 171:1630–1635
O’Neill LA (2008) The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunological reviews 226:10–18
Brown J, Wang H, Hajishengallis GN, Martin M (2011) TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. Journal of dental research 90:417–427
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature immunology 11:373–384
Estruch M, Bancells C, Beloki L, Sanchez-Quesada JL, Ordonez-Llanos J, Benitez S (2013) CD14 and TLR4 mediate cytokine release promoted by electronegative LDL in monocytes. Atherosclerosis 229:356–362
Sadanaga A, Nakashima H, Akahoshi M, Masutani K, Miyake K, Igawa T et al (2007) Protection against autoimmune nephritis in MyD88-deficient MRL/lpr mice. Arthritis and rheumatism 56:1618–1628
Matsushima N, Tanaka T, Enkhbayar P, Mikami T, Taga M, Yamada K et al (2007) Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC genomics 8:124
Wang Y, Ge P, Zhu Y (2013) TLR2 and TLR4 in the brain injury caused by cerebral ischemia and reperfusion. Mediators of inflammation, 2013:124614
Botos I, Segal DM, Davies DR (2011) The structural biology of Toll-like receptors. Structure (London, England : 1993) 19:447–459
Virtue A, Wang H, Yang XF (2012) MicroRNAs and toll-like receptor/interleukin-1 receptor signaling. Journal of hematology & oncology 5:66
Ciechomska M, Cant R, Finnigan J, van Laar JM, O’Reilly S (2013) Role of toll-like receptors in systemic sclerosis. Expert Reviews in Molecular Medicine 15:e9
Huang QQ, Pope RM (2009) The role of toll-like receptors in rheumatoid arthritis. Current Rheumatology Reports 11:357–364
Shi B, Huang Q, Tak PP, Vervoordeldonk MJ, Huang CC, Dorfleutner A et al (2012) SNAPIN: an endogenous Toll-like receptor ligand in rheumatoid arthritis. Annals of the Rheumatic Diseases 71:1411–1417
Richez C, Blanco P, Rifkin I, Moreau JF, Schaeverbeke T (2011) Role for toll-like receptors in autoimmune disease: the example of systemic lupus erythematosus. Joint, bone, spine : revue du rhumatisme 78:124–130
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650
Loiarro M, Volpe E, Ruggiero V, Gallo G, Furlan R, Maiorino C et al (2013) Mutational analysis identifies residues crucial for homodimerization of Myeloid Differentiation Factor 88 (MyD88) and for its function in immune cells. The Journal of Biological Chemistry
Lorne E, Dupont H, Abraham E (2010) Toll-like receptors 2 and 4: initiators of non-septic inflammation in critical care medicine? Intensive care medicine 36:1826–1835
Frazao JB, Errante PR, Condino-Neto A (2013) Toll-like receptors’ pathway disturbances are associated with increased susceptibility to infections in humans. Archivum immunologiae et therapiae experimentalis
Ori D, Kato H, Sanjo H, Tartey S, Mino T, Akira S et al (2013) Essential roles of K63-linked polyubiquitin-binding proteins TAB2 and TAB3 in B cell activation via MAPKs. Journal of Immunology (Baltimore, Md : 1950) 190:4037–4045
Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A et al (2009) Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 461:114–119
Clark K, Nanda S, Cohen P (2013) Molecular control of the NEMO family of ubiquitin-binding proteins. Nature Reviews Cancer 13:673–685
Qian C, Cao X (2013) Regulation of Toll-like receptor signaling pathways in innate immune responses. Ann N Y Acad Sci 1283:67–74
Picard C, Casanova JL, Puel A (2011) Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clinical Microbiology Reviews 24:490–497
Geurts J, van den Brand BT, Wolf A, Abdollahi-Roodsaz S, Arntz OJ, Kracht M et al (2011) Toll-like receptor 4 signalling is specifically TGF-beta-activated kinase 1 independent in synovial fibroblasts. Rheumatology (Oxford) 50:1216–1225
Ve T, Gay NJ, Mansell A, Kobe B, Kellie S (2012) Adaptors in toll-like receptor signaling and their potential as therapeutic targets. Current Drug Targets 13:1360–1374
Yang Y, Liao B, Wang S, Yan B, Jin Y, Shu HB et al (2013) E3 ligase WWP2 negatively regulates TLR3-mediated innate immune response by targeting TRIF for ubiquitination and degradation. Proc Natl Acad Sci U S A 110:5115–5120
Zhang M, Wang L, Zhao X, Zhao K, Meng H, Zhao W et al (2012) TRAF-interacting protein (TRIP) negatively regulates IFN-beta production and antiviral response by promoting proteasomal degradation of TANK-binding kinase 1. The Journal of experimental medicine 209:1703–1711
Sethurathinam S, Singh LP, Panneerselvam P, Byrne B, Ding JL (2013) UXT plays dual opposing roles on SARM-induced apoptosis. FEBS Letters 587:3296–3302
Perkins DJ, Polumuri SK, Pennini ME, Lai W, Xie P, Vogel SN (2013) Reprogramming of murine macrophages through TLR2 confers viral resistance via TRAF3-mediated, enhanced interferon production. PLoS Pathogens 9:e1003479
Jeong E, Lee JY (2011) Intrinsic and extrinsic regulation of innate immune receptors. Yonsei Medical Journal 52:379–392
Trengove MC, Ward AC (2013) SOCS proteins in development and disease. American Journal of Clinical and Experimental Immunology 2:1–29
Ahmed S, Maratha A, Butt AQ, Shevlin E, Miggin SM (2013) TRIF-mediated TLR3 and TLR4 signaling is negatively regulated by ADAM15. Journal of Immunology (Baltimore, Md : 1950) 190:2217–2228
Shio MT, Hassani K, Isnard A, Ralph B, Contreras I, Gomez MA et al (2012) Host cell signalling and leishmania mechanisms of evasion. Journal of Tropical Medicine 2012:819512
Kim EJ, Lee SM, Suk K, Lee WH (2012) CD300a and CD300f differentially regulate the MyD88 and TRIF-mediated TLR signalling pathways through activation of SHP-1 and/or SHP-2 in human monocytic cell lines. Immunology 135:226–235
Kim EJ, Suk K, Lee WH et al (2013) SHPS-1 and a synthetic peptide representing its ITIM inhibit the MyD88, but not TRIF, pathway of TLR signaling through activation of SHP and PI3K in THP-1 cells. Inflammation Research : Official Journal of the European Histamine Research Society 62:377–386
Sung NY, Yang MS, Song DS, Kim JK, Park JH, Song BS et al (2013) Procyanidin dimer B2-mediated IRAK-M induction negatively regulates TLR4 signaling in macrophages. Biochemical and Biophysical Research Communications 438:122–128
Lee HJ, Chung KC (2012) PINK1 positively regulates IL-1beta-mediated signaling through Tollip and IRAK1 modulation. Journal of Neuroinflammation 9:271
Srivastav S, Kar S, Chande AG, Mukhopadhyaya R, Das PK (2012) Leishmania donovani exploits host deubiquitinating enzyme A20, a negative regulator of TLR signaling, to subvert host immune response. Journal of Immunology (Baltimore, Md : 1950) 189:924–934
Zhong B, Liu X, Wang X, Liu X, Li H, Darnay BG et al (2013) Ubiquitin-specific protease 25 regulates TLR4-dependent innate immune responses through deubiquitination of the adaptor protein TRAF3. Science Signaling 6:ra35
Lin YC, Huang DY, Chu CL, Lin YL, Lin WW (2013) The tyrosine kinase Syk differentially regulates Toll-like receptor signaling downstream of the adaptor molecules TRAF6 and TRAF3. Science Signaling 6:ra71
Chuang TH, Ulevitch RJ (2004) Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nature immunology 5:495–502
Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L et al (2009) The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathogens 5:e1000650
Goh FG, Midwood KS (2012) Intrinsic danger: activation of Toll-like receptors in rheumatoid arthritis. Rheumatology (Oxford) 51:7–23
Takagi M (2011) Toll-like receptor—a potent driving force behind rheumatoid arthritis. Journal of Clinical and Experimental Hematopathology : JCEH 51:77–92
Neumann E, Lefevre S, Zimmermann B, Gay S, Muller-Ladner U (2010) Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends in Molecular Medicine 16:458–468
Ospelt C, Brentano F, Rengel Y, Stanczyk J, Kolling C, Tak PP et al (2008) Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis and Rheumatism 58:3684–3692
Radstake TR, Roelofs MF, Jenniskens YM, Oppers-Walgreen B, van Riel PL, Barrera P et al (2004) Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis and Rheumatism 50:3856–3865
Kowalski ML, Wolska A, Grzegorczyk J, Hilt J, Jarzebska M, Drobniewski M et al (2008) Increased responsiveness to toll-like receptor 4 stimulation in peripheral blood mononuclear cells from patients with recent onset rheumatoid arthritis. Mediators of Inflammation 2008:132732
Saber T, Veale DJ, Balogh E, McCormick J, NicAnUltaigh S, Connolly M et al (2011) Toll-like receptor 2 induced angiogenesis and invasion is mediated through the Tie2 signalling pathway in rheumatoid arthritis. PloS one 6:e23540
Chovanova L, Vlcek M, Krskova K, Penesova A, Radikova Z, Rovensky J et al (2013) Increased production of IL-6 and IL-17 in lipopolysaccharide-stimulated peripheral mononuclears from patients with rheumatoid arthritis. General Physiology and Biophysics 32:395–404
Tang CH, Hsu CJ, Yang WH, Fong YC (2010) Lipoteichoic acid enhances IL-6 production in human synovial fibroblasts via TLR2 receptor, PKCdelta and c-Src dependent pathways. Biochemical Pharmacology 79:1648–1657
Lorenz W, Buhrmann C, Mobasheri A, Lueders C, Shakibaei M (2013) Bacterial lipopolysaccharides form procollagen-endotoxin complexes that trigger cartilage inflammation and degeneration: implications for the development of rheumatoid arthritis. Arthritis Research & Therapy 15:R111
Chen Y, Sun W, Gao R, Su Y, Umehara H, Dong L et al (2013) The role of high mobility group box chromosomal protein 1 in rheumatoid arthritis. Rheumatology (Oxford) 52:1739–1747
Hu F, Mu R, Zhu J, Shi L, Li Y, Liu X et al. (2013) Hypoxia and hypoxia-inducible factor-1alpha provoke toll-like receptor signalling-induced inflammation in rheumatoid arthritis. Annals of the rheumatic diseases
Luo XJ, Mo XR, Zhou LL (2013) [The role of TLR2/4 in the IL-10 expression in synoviocytes of rheumatoid arthritis induced by Hsp72]. Zhongguo ying yong sheng li xue za zhi = Zhongguo yingyong shenglixue zazhi = Chinese journal of applied physiology, 29:212–3, 8
Huang QQ, Koessler RE, Birkett R, Dorfleutner A, Perlman H, Haines GK 3rd et al (2012) Glycoprotein 96 perpetuates the persistent inflammation of rheumatoid arthritis. Arthritis and rheumatism 64:3638–3648
Wahamaa H, Schierbeck H, Hreggvidsdottir HS, Palmblad K, Aveberger AC, Andersson U et al (2011) High mobility group box protein 1 in complex with lipopolysaccharide or IL-1 promotes an increased inflammatory phenotype in synovial fibroblasts. Arthritis research & therapy 13:R136
He Z, Shotorbani SS, Jiao Z, Su Z, Tong J, Liu Y et al (2012) HMGB1 promotes the differentiation of Th17 via up-regulating TLR2 and IL-23 of CD14+ monocytes from patients with rheumatoid arthritis. Scandinavian journal of immunology 76:483–490
Campo GM, Avenoso A, D’Ascola A, Prestipino V, Scuruchi M, Nastasi G et al (2012) Hyaluronan differently modulates TLR-4 and the inflammatory response in mouse chondrocytes. BioFactors (Oxford, England) 38:69–76
Abdollahi-Roodsaz S, Joosten LA, Koenders MI, Devesa I, Roelofs MF, Radstake TR et al (2008) Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. The Journal of clinical investigation 118:205–216
Hou Y, Lin H, Zhu L, Liu Z, Hu F, Shi J et al. (2013) LPS increases the incidence of collagen-induced arthritis in mice through induction of protease HTRA1 expression. Arthritis and rheumatism
Pierer M, Wagner U, Rossol M, Ibrahim S (2011) Toll-like receptor 4 is involved in inflammatory and joint destructive pathways in collagen-induced arthritis in DBA1J mice. PloS one 6:e23539
Ohto U, Yamakawa N, Akashi-Takamura S, Miyake K, Shimizu T (2012) Structural analyses of human Toll-like receptor 4 polymorphisms D299G and T399I. The Journal of biological chemistry 287:40611–40617
Emonts M, Hazes MJ, Houwing-Duistermaat JJ, van der Gaast-de Jongh CE, de Vogel L, Han HK et al. (2011) Polymorphisms in genes controlling inflammation and tissue repair in rheumatoid arthritis: a case control study. BMC medical genetics,12:36
Radstake TR, Franke B, Hanssen S, Netea MG, Welsing P, Barrera P et al (2004) The Toll-like receptor 4 Asp299Gly functional variant is associated with decreased rheumatoid arthritis disease susceptibility but does not influence disease severity and/or outcome. Arthritis and rheumatism 50:999–1001
Lee YH, Bae SC, Kim JH, Song GG (2013) Toll-like receptor polymorphisms and rheumatoid arthritis: a systematic review. Rheumatology International
Xu WD, Liu SS, Pan HF, Ye DQ (2012) Lack of association of TLR4 polymorphisms with susceptibility to rheumatoid arthritis and ankylosing spondylitis: a meta-analysis. Joint, bone, spine: revue du rhumatisme 79:566–569
Yang H, Wei C, Li Q, Shou T, Yang Y, Xiao C et al (2013) Association of TLR4 gene non-missense single nucleotide polymorphisms with rheumatoid arthritis in Chinese Han population. Rheumatology International 33:1283–1288
Han EC (2012) Systemic lupus erythematosus. The New England Journal of Medicine 366:573–574, author reply 4
Conti G, Coppo R, Amore A (2012) Pathogenesis of systemic lupus erythematosus (LES)]. Giornale italiano di nefrologia : organo ufficiale della Societa italiana di nefrologia 29(Suppl 54):S84–S90
Jallouli M, Frigui M, Marzouk S, Maaloul I, Kaddour N, Bahloul Z (2008) Infectious complications in systemic lupus erythematosus: a series of 146 patients. La Revue de medecine interne/fondee par la Societe nationale francaise de medecine interne 29:626–631
Kirchner M, Sonnenschein A, Schoofs S, Schmidtke P, Umlauf VN, Mannhardt-Laakmann W (2013) Surface expression and genotypes of Toll-like receptors 2 and 4 in patients with juvenile idiopathic arthritis and systemic lupus erythematosus. Pediatric Rheumatology Online Journal 11:9
Komatsuda A, Wakui H, Iwamoto K, Ozawa M, Togashi M, Masai R et al (2008) Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clinical and experimental immunology 152:482–487
Fujita K, Akasaka Y, Kuwabara T, Wang B, Tanaka K, Kamata I et al (2012) Pathogenesis of lupus-like nephritis through autoimmune antibody produced by CD180-negative B lymphocytes in NZBWF1 mouse. Immunology Letters 144:1–6
Tsao JT, Hsieh SC, Chiang BL, Yu CL, Lin SC (2012) Altered IL-10 and TNF-alpha production in peripheral blood mononuclear cells of systemic lupus erythematosus patients after Toll-like receptor 2, 4, or 9 activation. Clinical and experimental medicine 12:153–158
Wen Z, Xu L, Chen X, Xu W, Yin Z, Gao X et al (2013) Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway. Journal of Immunology (Baltimore, Md: 1950) 190:5411–5422
Loser K, Vogl T, Voskort M, Lueken A, Kupas V, Nacken W et al (2010) The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nature Medicine 16:713–717
Lartigue A, Colliou N, Calbo S, Francois A, Jacquot S, Arnoult C et al (2009) Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. Journal of Immunology (Baltimore, Md : 1950) 183:6207–6216
Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J et al (2010) The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. The Journal of Clinical Investigation 120:4251–4272
Lee TP, Tang SJ, Wu MF, Song YC, Yu CL, Sun KH (2010) Transgenic overexpression of anti-double-stranded DNA autoantibody and activation of Toll-like receptor 4 in mice induce severe systemic lupus erythematosus syndromes. Journal of Autoimmunity 35:358–367
Summers SA, Hoi A, Steinmetz OM, O’Sullivan KM, Ooi JD, Odobasic D et al (2010) TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. Journal of Autoimmunity 35:291–298
Castiblanco J, Varela DC, Castano-Rodriguez N, Rojas-Villarraga A, Hincapie ME, Anaya JM (2008) TIRAP (MAL) S180L polymorphism is a common protective factor against developing tuberculosis and systemic lupus erythematosus. Infection, Genetics and Evolution : Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases 8:541–544
Wu M, Assassi S (2013) The role of type 1 interferon in systemic sclerosis. Frontiers in Immunology 4:266
Fuschiotti P, Larregina AT, Ho J, Feghali-Bostwick C, Medsger TA Jr (2013) Interleukin-13-producing CD8+ T cells mediate dermal fibrosis in patients with systemic sclerosis. Arthritis and Rheumatism 65:236–246
van Lieshout AW, Vonk MC, Bredie SJ, Joosten LB, Netea MG, van Riel PL et al (2009) Enhanced interleukin-10 production by dendritic cells upon stimulation with Toll-like receptor 4 agonists in systemic sclerosis that is possibly implicated in CCL18 secretion. Scandinavian Journal of Rheumatology 38:282–290
van Bon L, Popa C, Huijbens R, Vonk M, York M, Simms R et al (2010) Distinct evolution of TLR-mediated dendritic cell cytokine secretion in patients with limited and diffuse cutaneous systemic sclerosis. Annals of the Rheumatic Diseases 69:1539–1547
Fineschi S, Goffin L, Rezzonico R, Cozzi F, Dayer JM, Meroni PL et al (2008) Antifibroblast antibodies in systemic sclerosis induce fibroblasts to produce profibrotic chemokines, with partial exploitation of toll-like receptor 4. Arthritis and Rheumatism 58:3913–3923
Bhattacharyya S, Kelley K, Melichian DS, Tamaki Z, Fang F, Su Y et al (2013) Toll-like receptor 4 signaling augments transforming growth factor-beta responses: a novel mechanism for maintaining and amplifying fibrosis in scleroderma. The American Journal of Pathology 182:192–205
Wermuth PJ, Jimenez SA (2012) Gadolinium compounds signaling through TLR4 and TLR7 in normal human macrophages: establishment of a proinflammatory phenotype and implications for the pathogenesis of nephrogenic systemic fibrosis. Journal of Immunology (Baltimore, Md : 1950 189:318–327
Yoshizaki A, Iwata Y, Komura K, Ogawa F, Hara T, Muroi E et al (2008) CD19 regulates skin and lung fibrosis via Toll-like receptor signaling in a model of bleomycin-induced scleroderma. The American Journal of Pathology 172:1650–1663
Vakaloglou KM, Mavragani CP (2011) Activation of the type I interferon pathway in primary Sjogren’s syndrome: an update. Current Opinion in Rheumatology 23:459–464
Nguyen CQ, Peck AB (2013) The interferon-signature of Sjogren’s syndrome: how unique biomarkers can identify underlying inflammatory and immunopathological mechanisms of specific diseases. Frontiers in Immunology 4:142
Kawakami A, Nakashima K, Tamai M, Nakamura H, Iwanaga N, Fujikawa K et al (2007) Toll-like receptor in salivary glands from patients with Sjogren’s syndrome: functional analysis by human salivary gland cell line. The Journal of Rheumatology 34:1019–1026
Kwok SK, Cho ML, Her YM, Oh HJ, Park MK, Lee SY et al (2012) TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in patients with primary Sjogren’s syndrome. Arthritis Research & Therapy 14:R64
Spachidou MP, Bourazopoulou E, Maratheftis CI, Kapsogeorgou EK, Moutsopoulos HM, Tzioufas AG et al (2007) Expression of functional Toll-like receptors by salivary gland epithelial cells: increased mRNA expression in cells derived from patients with primary Sjogren’s syndrome. Clinical and Experimental Immunology 147:497–503
Chong HT, Kopecki Z, Cowin AJ (2013) Lifting the silver flakes: the pathogenesis and management of chronic plaque psoriasis. BioMed Research International 2013:168321
Carrasco S, Neves FS, Fonseca MH, Goncalves CR, Saad CG, Sampaio-Barros PD et al (2011) Toll-like receptor (TLR) 2 is upregulated on peripheral blood monocytes of patients with psoriatic arthritis: a role for a gram-positive inflammatory trigger? Clinical and Experimental Rheumatology 29:958–962
Garcia-Rodriguez S, Arias-Santiago S, Perandres-Lopez R, Castellote L, Zumaquero E, Navarro P et al (2013) Increased gene expression of Toll-like receptor 4 on peripheral blood mononuclear cells in patients with psoriasis. Journal of the European Academy of Dermatology and Venereology : JEADV 27:242–250
Castrop F, Haslinger B, Hemmer B, Buck D (2013) Review of the pharmacoeconomics of early treatment of multiple sclerosis using interferon beta. Neuropsychiatric Disease and Treatment 9:1339–1349
Shaw PJ, Barr MJ, Lukens JR, McGargill MA, Chi H, Mak TW et al (2011) Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity 34:75–84
Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T (2010) Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A 107:11555–11560
Andersson A, Covacu R, Sunnemark D, Danilov AI, Dal Bianco A, Khademi M et al (2008) Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. Journal of Leukocyte Biology 84:1248–1255
Reynolds JM, Martinez GJ, Chung Y, Dong C (2012) Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci U S A 109:13064–13069
Bustamante MF, Fissolo N, Rio J, Espejo C, Costa C, Mansilla MJ et al (2011) Implication of the Toll-like receptor 4 pathway in the response to interferon-beta in multiple sclerosis. Annals of Neurology 70:634–645
Nokoff N, Rewers M (2013) Pathogenesis of type 1 diabetes: lessons from natural history studies of high-risk individuals. Ann N Y Acad Sci 1281:1–15
Lien E, Zipris D (2009) The role of Toll-like receptor pathways in the mechanism of type 1 diabetes. Current Molecular Medicine 9:52–68
Devaraj S, Jialal I, Yun JM, Bremer A (2011) Demonstration of increased toll-like receptor 2 and toll-like receptor 4 expression in monocytes of type 1 diabetes mellitus patients with microvascular complications. Metabolism: Clinical and Experimental 60:256–259
Ururahy MA, Loureiro MB, Freire-Neto FP, de Souza KS, Zuhl I, Brandao-Neto J et al (2012) Increased TLR2 expression in patients with type 1 diabetes: evidenced risk of microalbuminuria. Pediatric Diabetes 13:147–154
Li M, Song L, Gao X, Chang W, Qin X (2012) Toll-like receptor 4 on islet beta cells senses expression changes in high-mobility group box 1 and contributes to the initiation of type 1 diabetes. Experimental & Molecular Medicine 44:260–267
Jagannathan M, McDonnell M, Liang Y, Hasturk H, Hetzel J, Rubin D et al (2010) Toll-like receptors regulate B cell cytokine production in patients with diabetes. Diabetologia 53:1461–1471
Yin J, Peng Y, Wu J, Wang Y, Yao L (2013) Toll-like receptor 2/4 links to free fatty acid-induced inflammation and beta-cell dysfunction. Journal of Leukocyte Biology
Dong B, Qi D, Yang L, Huang Y, Xiao X, Tai N et al (2012) TLR4 regulates cardiac lipid accumulation and diabetic heart disease in the nonobese diabetic mouse model of type 1 diabetes. American journal of Physiology Heart and Circulatory Physiology 303:H732–H742
Devaraj S, Tobias P, Jialal I (2011) Knockout of toll-like receptor-4 attenuates the pro-inflammatory state of diabetes. Cytokine 55:441–445
Devaraj S, Tobias P, Kasinath BS, Ramsamooj R, Afify A, Jialal I (2011) Knockout of toll-like receptor-2 attenuates both the proinflammatory state of diabetes and incipient diabetic nephropathy. Arteriosclerosis, Thrombosis, and Vascular Biology 31:1796–1804
Lee MS, Kim DH, Lee JC, Kim S, Kim HS (2011) Role of TLR2 in the pathogenesis of autoimmune diabetes and its therapeutic implication. Diabetes/Metabolism Research and Reviews 27:797–801
Filippi CM, Ehrhardt K, Estes EA, Larsson P, Oldham JE, von Herrath MG (2011) TLR2 signaling improves immunoregulation to prevent type 1 diabetes. European Journal of Immunology 41:1399–1409
Kim DH, Lee JC, Kim S, Oh SH, Lee MK, Kim KW et al (2011) Inhibition of autoimmune diabetes by TLR2 tolerance. Journal of Immunology (Baltimore, Md : 1950) 187:5211–5220
Li J, Wang X, Zhang F, Yin H (2013) Toll-like receptors as therapeutic targets for autoimmune connective tissue diseases. Pharmacology & Therapeutics 138:441–451
Li G, Liu D, Zhang Y, Qian Y, Zhang H, Guo S et al (2013) Celastrol inhibits lipopolysaccharide-stimulated rheumatoid fibroblast-like synoviocyte invasion through suppression of TLR4/NF-kappaB-mediated matrix metalloproteinase-9 expression. PloS One 8:e68905
Wang J, Lu J, Lan Y, Zhou H, Li W, Xiang M (2013) Total coumarins from Urtica dentata Hand prevent murine autoimmune diabetes via suppression of the TLR4-signaling pathways. Journal of Ethnopharmacology 146:379–392
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 81220108017, no. 81373205, and no. 81270024), the Specialized Research Fund for the Doctoral Program of Higher Education (grant no. 20120162130003), and the Programs of Science-Technology Commission of Hunan province (2013FJ4202, 2011TP4019-7) and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, Y., Yin, H., Zhao, M. et al. TLR2 and TLR4 in Autoimmune Diseases: a Comprehensive Review. Clinic Rev Allerg Immunol 47, 136–147 (2014). https://doi.org/10.1007/s12016-013-8402-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-013-8402-y