
Abstract
Congenital neutropenia syndromes comprise a heterogeneous group of disorders leading to increased susceptibility to bacterial infections. Recent work has elucidated the molecular basis of several congenital neutropenia syndromes such as mutations in ELA2, HAX1, GF11, and WAS. In addition, a number of complex clinical syndromes associating congenital neutropenia have been recognized and elucidated on a genetic level, e.g. p14-deficiency or G6PC3-deficiency. The clinical and genetic findings of various neutropenia syndromes are being discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Severe congenital neutropenia (SCN), originally described by Rolf Kostmann in 1950 [1], is a primary immunodeficiency syndrome characterized by early onset of severe bacterial infections. Due to a paucity of peripheral neutrophil granulocytes (<500/µl) patients are prone to invasive infections, typically in the absence of pus formation. “Infantile agranulocytosis”, as Rolf Kostmann named this hereditary syndrome, has been known for more than half a century, yet the underlying genetic mutations have remained unknown for many decades.
Modern genetic research in the field of congenital neutropenia has not only allowed a rational approach to classifying SCN but has also shed light on molecular mechanisms controlling the development and function of neutrophil granulocytes.
From a clinical point of view, two major groups of inherited immunodeficiency diseases associated with congenital neutropenia can be distinguished, first, severe congenital neutropenia without additional organ manifestations, and second, severe congenital neutropenia associated with syndromal features [2].
Here, we provide an overview of recent progress in identifying genetic defects causing congenital neutropenia.
-
(a)
Congenital neutropenia without syndromal features
Severe congenital neutropenia
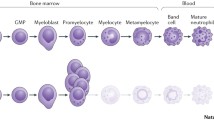
In contrast to cyclic neutropenia, which characterized by oscillating neutrophil counts, SCN classically shows continuously low neutrophil counts. The typical morphological finding is a “myeloid maturation arrest” at the level of promyelocytes, seen in conventional bone marrow smears.
Using genetic linkage studies, Horwitz and colleagues defined mutations in the gene encoding neutrophil elastase (ELA2) as cause of autosomal dominant cyclic neutropenia [3]. Subsequently, sporadic and autosomal dominant cases of SCN have also been associated with ELA2 mutations (reviewed in [4]), suggesting that the feature of cycling hematopoiesis may not be a strong discriminatory factor defining subgroups of neutropenia disorders. Even though certain ELA2 mutations appear to be seen more frequently in patients with cyclic neutropenia, it remains unclear, why some ELA2 variants are associated with cyclic neutropenia while others are associated with SCN (Table 1).
ELA2 mutations are either transmitted in an autosomal dominant inheritance pattern or occur sporadically. Recently, Boxer and co-workers published compelling data supporting autosomal dominant transmission of ELA2 mutations as cause for SCN. Five unrelated children, all affected by severe congenital neutropenia, were found to be descendents of a single sperm donor carrying an ELA2 mutation, whose semen was used to impregnate five healthy mothers [5]. A few patients with mosaicism have also been reported [6, 7].
In Caucasian patients, ELA2 mutations appear to represent the most common genetic variant causing SCN, accounting for approximately 50–60% of SCN [8, 9]. Neutrophil elastase is a serine protease exclusively expressed in neutrophils and monocytes and displays pleiotropic functions. It has direct antimicrobial activity and also integrates inflammatory responses by proteolytic modification of chemokines, cytokines, and cell surface receptors (reviewed in [10]).
It is still unclear, how heterozygous mutations in ELA2 cause SCN. Benson suggested that aberrant intracellular trafficking of mutated neutrophil elastase may explain non-physiological enzymatic activity ultimately leading to neutropenia [11]. Alternatively, mutated elastase may be misprocessed and degraded via the activation of the so-called unfolded protein response [12]. For a comprehensive review, the reader is referred to an excellent summary on this topic [4].
Few SCN patients have been described who have mutations in WAS [13], the gene encoding the Wiskott–Aldrich-Syndrome protein. While patients with Wiskott–Aldrich syndrome have loss of function mutations in WAS [14], patients with congenital neutropenia may have gain of function mutations. Physiologically, inadequate activation of WASP is controlled by a conformational change leading to autoinhibition. Certain mutations in WAS prevent this autoinhibitory conformation and thus lead to excessive actin polymerisation. This altered state of the actin cytoskeleton also implies aberrant mitosis and may lead to chromosomal instability and myelodysplastic features [15].
It remains controversial whether the phenotype of myeloid maturation arrest may be caused by increased cell death of mature neutrophils and/or aberrant myeloid differentiation. Since differentiation is controlled by transcription factors, a search for dysfunctional transcription factors involved in SCN has been initiated by several groups. GFI1 is a transcriptional repressor and splice-control factor of the zinc-finger family of transcription factors and has been implicated in the differentiation of various hematopoietic cell lineages, including neutrophils [16, 17]. Two families with heterozygous mutations in GFI1 and congenital neutropenia have been described [18]. GFI1 mutations act in a dominant-negative way and cause dysregulation of several target genes such as C/EBPepsilon [19], ELA2 [18], and the monopoietic cytokine Csf1 [20].
Further evidence for aberrant transcriptional control of neutrophil development in SCN patients stems from an analysis of the Wnt signaling pathway. Wnt proteins are involved in mitogenic stimulation, cell fate specification, and differentiation. In unstimulated cells the transcriptional regulator β-catenin is degraded via a complex interplay of factors, such as disheveled, glycogen synthase kinase-3β, Axin, and adenomatous polyposis coli. In the so-called canonical Wnt pathway, Wnt proteins bind to their receptor, the frizzled/low density lipoprotein receptor-related protein complex, and inhibit the degradation of β-catenin. β-catenin accumulates in the cytoplasm and nucleus where it regulates transcription through interaction with the lymphoid enhancer factor (LEF-) 1 (reviewed in [21]). Skokowa and co-workers provided evidence that in myeloid progenitor cells from Kostmann-type SCN patients LEF-1 as well as LEF-1 target genes, e.g. cyclin D1, c-myc. ELA2, surviving, and C/EBPα are transcriptionally downregulated [22]. Further mechanistic studies will have to show how decreased LEF-1 may inhibit neutrophil differentiation.
While ELA2 and GFI1 may account for sporadic or autosomal dominant variants of SCN, the underlying molecular genetic defect of classical, autosomal recessive Kostmann-type SCN remained enigmatic more than 50 years after the first description (reviewed in [23]). Using linkage analysis and a candidate gene sequencing approach, we have identified homozygous mutations in the antiapoptotic molecule HAX1 in patients with autosomal recessive SCN [24]. HAX1 is known to interfere with signal transduction and cytoskeletal control [25–27]. Furthermore, HAX1 is a critical protein for maintaining the inner mitochondrial membrane potential and protects myeloid cells from apoptosis [24]. Deficiency of this key protein destabilizes the inner mitochondrial membrane potential and leads to SCN via increased apoptosis of myeloid progenitors. At least two transcripts of HAX1 have been identified. Interestingly, some patients with HAX1 deficiency suffer from neurological problems, ranging from mild cognitive problems to severe developmental delay and epilepsy [28–31]. These differences in phenotype can be attributed to the type of mutation in HAX1, which has two distinct splice variants. In case the mutation affects only isoform A, patients suffer from congenital neutropenia but not from neurological problems. In case the mutation affects both isoform A and isoform B, all patients show signs and symptoms of neurological impairment, indicating a strict genotype–phenotype correlation [32, 33].
Interestingly, not all patients with autosomal recessive SCN have mutations in HAX1. Ongoing studies in our laboratories suggest that there are several additional genes involved in the pathogenesis of autosomal recessive SCN (unpublished). Despite the phenotypical similarities in patients with HAX1 deficiency and ELA2 mutations [34], no common mechanism has been characterized explaining why both of the defects lead to isolated congenital neutropenia.
-
(b)
Congenital neutropenia associated with syndromal features
A subset of patients with congenital neutropenia has additional features such as lymphoid cell dysfunction, pigmentation defects, inner ear hearing loss, or cardiac/urogenital anomalies. Elucidation of the underlying genetic and molecular defects has provided fascinating insights into the basic function of mitochondria, lysosomes, and the endoplasmic retriculum.
Reticular dysgenesis
Reticular dysgenesis is an autosomal recessive form of early differentiation arrest in the myeloid lineage associated with severe combined immunodeficiency due to impaired lymphoid development [35]. Furthermore, affected patients suffer from sensorineural hearing loss [36]. Congenital neutropenia in reticular dysgenesis is non-responsive to granulocyte colony stimulating factor (G-CSF) [37]. Using a genome linkage analysis and candidate gene sequencing approach, mutations in adenylate kinase 2 (AK2) have been identified by two independent groups [38, 39]. Like HAX1, AK2 is localized in the mitochondrial intermembrane space and may be important in mitochondrial energy metabolism and control of apoptosis via FADD and caspase 10 [40].
Congenital neutropenia with hypopigmentation
While Chédiak-Higashi syndrome (CHS) [41, 42] and Griscelli syndrome type 2 (GS2) [43] may only show transient episodes of neutropenia, Hermansky–Pudlak syndrome type 2 (HPS2) and p14 deficiency are characterized by constantly reduced peripheral neutrophil counts [44, 45]. All four disorders can be differentiated based on their clinical phenotype [46]. In contrast to SCN with myeloid maturation arrest, CHS, GS2, HPS2, and p14 deficiency show presence of mature neutrophils in the bone marrow.
Hermansky–Pudlak syndrome was originally described as oculocutaneous albinism and platelet storage disease with increased bleeding tendency [41, 42]. Currently, HPS comprises a group of at least eight distinct genetic defects in humans (reviewed in [47]). All defective genes play a role in intracellular protein trafficking. As a result of the mutations, dysfunction of lysosome-related organelles (LROs) is seen in cells from HPS patients.
Among all variants of HPS, only HPS2 is characterized by congenital neutropenia [48–50]. The first patients, two descendents of a consanguineous pedigree, were described by Kotzot and colleagues in 1994 [51]. Up to date, less than ten patients have been described in the literature [44, 48–55]. HPS2 is caused by mutations in the AP3B1 gene which encodes the β3A subunit of the heterotetrameric adaptor protein (AP-) 3 complex. β3A deficiency leads to an almost complete abolishment of the remaining AP-3 subunits, i.e. δ, μ3A and σ3A. The AP-3 complex mediates cargo protein selection into transport vesicles and their trafficking to lysosomes [53, 56]. In the absence of AP-3, proteins destined to traffic to lysosomes therefore accumulate at the plasma membrane. It remains unknown why patients with AP-3 deficiency are neutropenic. So far, no functional defect could be identified in AP-3-deficient neutrophils [44]. Interestingly, AP-3-deficient dogs show cyclic neutropenia and AP-3-deficient mice appear to have no neutrophil defect (reviewed in [47]). However, AP-3 deficiency is associated with defects of NK and NKT cells [44, 55]. Lethal hemophagocytosis has recently been reported in one patient [52], suggesting that defective cytotoxicity may be clinically relevant in AP-3 deficiency.
A previously unrecognized immunodeficiency syndrome associating growth delay, partial albinism, lymphoid deficiency, and congenital neutropenia has recently been described in four members of a large pedigree [45]. Using a combination approach of genome wide linkage analysis and transcriptional profiling, we identified a homozygous mutation in the gene encoding the endosomal adaptor molecule p14. A point mutation in the 3’-UTR leads to decreased p14 mRNA. As a consequence of decreased p14 protein levels, neutrophils show an altered ultrastructure of azurophilic granules and decreased microbicidal activity in phagosomes. Furthermore, p14-deficient cells have a defect in cytokine-receptor-mediated ERK phosphorylation and show a marked dislocalisation of late endosomes. p14 is ubiquitously expressed and located at the outer membrane of late endosomes [57, 58]. The molecule acts as a scaffold protein by interacting with MEK partner (MP) 1 to direct the mitogen-activated protein kinase pathway. Its function is crucial for subcellular compartmentalization of signals [59]. Recently, a murine p14-deficient mouse model has been generated [60]. Complete knock-out of the p14 gene causes early intrauterine lethality. Epidermis-specific knock-out of p14 in mice results in a severely reduced number of epidermal skin layers, perturbed terminal skin differentiation, aberrant epidermal growth factor receptor degradation, and defective cellular proliferation. Affected mice die shortly after birth [60]. These studies support the idea that p14 is critical for cellular proliferation and differentiation. p14-deficient patients may only survive because of residual p14 protein levels (hypomorphic phenotype). Of note, some patients with a phenotype of p14 deficiency are known who do not show any mutation in p14, suggesting that this disorder is heterogeneous.
Congenital neutropenia and glycogen metabolism
Neutrophils are exquisitely sensitive to glucose deprivation. Two human genetic disorders have been identified in which neutrophil granulocytes show a predisposition to accelerated apoptosis due to disturbances of energy metabolism in the endoplasmic reticulum. Patients with glycogenosis type 1b, a variant of glycogenosis type 1, is not only characterized by glycogen storage, hypoglycaemia, and lactic acidosis, but also show a paucity of mature neutrophil granulocytes. This disease is caused by mutations in the ubiquitously expressed glucose-6-phosphate translocase (G6PT, encoded by the gene SLC37A4), a transporter mediating translocation of G6P into the endoplasmic reticulum. G6PT-deficient neutrophils show a significantly impaired respiratory-burst response. Therefore, GSD1b patients are not only at risk for severe bacterial infections due to neutropenia but may also develop clinical symptoms secondary to poor neutrophil function, such as inflammatory bowel disease [61].
Recently, we identified a second congenital neutropenia syndrome associated with defective glucose metabolism. In contrast to GSD1 and GSD1b, affected patients do not show signs of glycogen storage or symptoms related to hypoglycemia. Instead, there is an association of congenital neutropenia, various developmental defects of the cardiovascular and/or urogenital system and increased visibility of superficial veins [62]. There is a high degree of variation in clinical presentation and the determinants of the wide clinical spectrum remain unknown at the present time. Cardiac anomalies may include atrial septal defect, cor triatriatum or pulmonary vein stenosis, and urogenital problems comprise urachal fistulation, renal dysgenesis, or undescended testes. Myeloid progenitor cells are characterized by increased signs of the “unfolded protein response” or ER-stress, as evidenced by ultrastructural anomalies and increased expression of the chaperone BiP [62]. The defective protein, G6PC3, is a paralog of G6PC1, the classical glucose-6-phosphate, mutated in GSD1. In contrast to G6PC1, G6PC3 is expressed ubiquitously, suggesting that G6PC3 may in fact be the phylogenetically more ancient variant of glucose-6-phosphatase. G6PC2 is expressed in the pancreatic islets and has recently been implicated in diabetes mellitus [63, 64]. In the absence of G6PC3, myeloid cells are prone to undergo apoptosis, and we speculate that aberrant apoptosis due to increased ER-stress may also be the cause for defective organ development.
WHIM syndrome (warts, hypogammaglobulinemia, immunodeficiency, myelokathexis)
Myelokathexis is defined as dysgranulopoiesis (hypersegmented neutrophils, increased apoptosis) with increased bone marrow cellularity. First described by Zuelzer in a sporadic case [65], myelokathexis was subsequently found to occur as an autosomal dominant trait in association with hypogammaglobulinemia and dermal/genital papilloma virus infections [66]. Severe infections are rare since neutrophils can be adequately released to the periphery in case of infections.
Hernandez and colleagues [67] identified heterozygous mutations in the chemokine receptor gene CXCR4 in WHIM syndrome patients. Mutant CXCR4 shows significantly increased calcium flux in response to its ligand stromal-derived factor-1α (SDF-1α) consistent with dysregulated signaling of the mutant receptor. Likewise, SDF-1α stimulation of neutrophils with a truncating CXCR4 mutation, but not with non-mutated CXCR4, blocked the constitutive apoptosis seen in neutrophils from WHIM patients [68]. Kawai and colleagues [69] investigated the role of CXCR4 mutations in myelokathexis. They transduced wild type and mutated CXCR4 in human CD34+ peripheral blood-mobilized stem cells (PBSCs) and analyzed the properties of these cells in cell culture and after transplantation into nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice, an elegant xenotransplantation system to unequivocally assess the engraftment and expansion of the transplanted human cells. NOD/SCID mice are devoid of B and T cells and have a defect in Natural Killer cell function. Thus, they do not reject the xenograft. Interestingly, mutant CXCR4 transduced PBSCs behaved like the wild-type CXCR4 transduced PBSCs in the cell culture system but showed a better engraftment with a significantly enhanced apoptosis and decreased release of mature neutrophils from the BM only after transplantation into NOD/SCID mice suggesting that the CXCR4 mutation alone is insufficient to explain the increased apoptosis of BM stem cells in WHIM patients.
Interestingly, not all patients with myelokathexis have mutations in CXCR4 [70]. In particular, the WHIM phenotype can also be transmitted as an autosomal recessive trait, suggesting that other genes are involved in this disorder.
Natural history, treatment, and complications
Before recombinant human granulocyte colony stimulating factor became available, most patients with SCN succumbed to severe and recurrent bacterial infections in infancy [70–72]. In the G-CSF era, morbidity and mortality from infectious complications has greatly decreased and the quality of life has markedly improved. Today, most patients with congenital neutropenia survive into adulthood.
G-CSF has pleiotropic effects by orchestrating a series of molecular events in favor of neutrophil differentiation. In addition to its effects on differentiation, G-CSF has antiapoptotic effects. A very recent study has shown that G-CSF acts through Nampt/PEBF to increase intracellular NAD1, which in combination with SIRT1 binds and activates C/EBP-beta and leads to the so-called emergency neutrophil granulopoies [73].
Long-term studies, facilitated by the international congenital neutropenia registries in Seattle, USA, and Hannover, Germany, revealed long-term complications. There is a small but significant risk of death secondary to infectious complications [74]. In spite of effective G-CSF treatment, there is a risk (0.9% per year) of SCN patients to die as a consequence of infectious complications [74]. Thus, the reconstitution of normal neutrophil counts may not suffice to re-establish antibacterial immunity. ELA2-mutated patients show abnormal expression of neutrophil elastase and other granule-associated proteins such as myeloperoxidase, lactoferrin, cathepsin-G, and human-neutrophil-peptide [75]. These abnormalities may contribute to altered neutrophil function. More importantly, many SCN patients develop clonal hematopoietic disorders such as myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML). After 12 consecutive years of treatment, the risk is 8% per year [74]. Long-term studies have not shown a plateau effect, so there is a certain likelihood that the overall risk of developing leukemia continues to rise. The molecular etiology of leukemogenesis remains unknown. Many SCN patients develop somatic mutations in the CSF3R gene. Germeshausen and colleagues followed up on CSF3R mutations in 148 SCN patients 23 of whom developed signs of malignant transformation [76]. Eighteen of these individuals had acquired CSF3R mutations. Additionally, in 50 patients analyzed they could show an accumulation of different CSF3R mutations over time.
CSF3R mutations appear in early hematopoietic progenitor cells and confer a survival advantage in myeloid cells [77]. This observation has also been seen in murine studies. Liu et al. [78] used a murine model in which HSC expressed a truncating mutation of the G-CSF-R. This study demonstrated that administration of G-CSF permitted the outgrowth of G-CSF-Rmut stem cells in competitive repopulation experiments. This selective advantage was associated with and dependent on G-CSF induced increased activation of STAT5. However, no autonomous G-CSF-independent growth was observed and none of the mice developed AML or MDS.
In spite of successful supportive treatment using recombinant human G-CSF, definitive and curative treatment of SCN is currently only possible by means of allogeneic bone marrow transplantation. Preclinical studies using transplantation of genetically corrected hematopoietic stem cells are under way, but no clinical protocol has been opened up to date.
Conclusions and perspectives
Suspicion of congenital neutropenia should prompt a complete diagnostic workup including molecular diagnosis. The identification of underlying genetic defects now allows a molecular diagnosis in many patients, a prerequisite for a better assessment of the natural evolution and therapy-related risks. Furthermore, genetic diagnosis is the first step towards development of gene-based therapies. However, in many patients with congenital neutropenia underlying mutations are still unknown. Therefore, sustained scientific efforts and multi-national collaborations are needed to elucidate the genetic basis of all remaining variants of SCN.
References
Kostmann R (1950) Hereditär reticulos-en ny systemsjukdom. Svenska Laekartidningen 47:2861–2868
Boztug K et al (2008) Congenital neutropenia syndromes. Immunol Allergy Clin North Am 28(2):259–275 vii-viii
Horwitz M et al (1999) Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nat Genet 23(4):433–436
Horwitz MS et al (2007) Neutrophil elastase in cyclic and severe congenital neutropenia. Blood 109(5):1817–1824
Boxer LA et al (2006) Strong evidence for autosomal dominant inheritance of severe congenital neutropenia associated with ELA2 mutations. J Pediatr 148(5):633–636
Ancliff PJ, Gale RE, Linch DC (2003) Neutrophil elastase mutations in congenital neutropenia. Hematology 8(3):165–171
Germeshausen M et al (2001) Mutations in the gene encoding neutrophil elastase (ELA2) are not sufficient to cause the phenotype of congenital neutropenia. Br J Haematol 115(1):222–224
Bellanne-Chantelot C et al (2004) Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood 103(11):4119–4125
Skokowa J et al (2007) Severe congenital neutropenia: inheritance and pathophysiology. Curr Opin Hematol 14(1):22–28
Pham CT (2006) Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 6(7):541–550
Benson KF et al (2003) Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet 35(1):90–96
Kollner I et al (2006) Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood 108(2):493–500
Devriendt K et al (2001) Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet 27(3):313–317
Thrasher AJ (2002) WASp in immune-system organization and function. Nat Rev Immunol 2(9):635–646
Moulding DA et al (2007) Unregulated actin polymerization by WASp causes defects of mitosis and cytokinesis in X-linked neutropenia. J Exp Med 204(9):2213–2224
Karsunky H et al (2002) Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat Genet 30(3):295–300
Hock H et al (2003) Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity 18(1):109–120
Person RE et al (2003) Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet 34(3):308–312
Zhuang D et al (2006) Increased CCAAT enhancer-binding protein epsilon (C/EBPepsilon) expression and premature apoptosis in myeloid cells expressing Gfi-1 N382S mutant associated with severe congenital neutropenia. J Biol Chem 281(16):10745–10751
Zarebski A et al (2008) Mutations in growth factor independent-1 associated with human neutropenia block murine granulopoiesis through colony stimulating factor-1. Immunity 28(3):370–380
Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810
Skokowa J et al (2006) LEF-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced in congenital neutropenia. Nat Med 12(10):1191–1197
Carlsson G et al (2006) Neutrophil elastase and granulocyte colony-stimulating factor receptor mutation analyses and leukemia evolution in severe congenital neutropenia patients belonging to the original Kostmann family in northern Sweden. Haematologica 91(5):589–595
Klein C et al (2007) HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet 39(1):86–92
Suzuki Y et al (1997) HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J Immunol 158(6):2736–2744
Gallagher AR et al (2000) The polycystic kidney disease protein PKD2 interacts with Hax-1, a protein associated with the actin cytoskeleton. Proc Natl Acad Sci U S A 97(8):4017–4022
Radhika V et al (2004) Galpha13 stimulates cell migration through cortactin-interacting protein Hax-1. J Biol Chem 279(47):49406–49413
Carlsson G, Fasth A (2001) Infantile genetic agranulocytosis, morbus Kostmann: presentation of six cases from the original "Kostmann family" and a review. Acta Paediatr 90(7):757–764
Rezaei N et al (2007) Association of HAX1 deficiency with neurological disorder. Neuropediatrics 38(5):261–263
Matsubara K et al (2007) Severe developmental delay and epilepsy in a Japanese patient with severe congenital neutropenia due to HAX1 deficiency. Haematologica 92(12):e123–e125
Ishikawa N et al (2008) Neurodevelopmental abnormalities associated with severe congenital neutropenia due to the R86X mutation in the HAX1 gene. J Med Genet 45(12):802–807
Germeshausen M et al (2008) Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood 111(10):4954–4957
Carlsson G et al (2008) Central nervous system involvement in severe congenital neutropenia: neurological and neuropsychological abnormalities associated with specific HAX1 mutations. J Intern Med 264(4):388–400
Zeidler C et al (2009) Clinical implications of ELA2-, HAX1-, and G-CSF-receptor (CSF3R) mutations in severe congenital neutropenia. Br J Haematol 144(4):459–467
de VO, Seynhaeve V (1959) Reticular dysgenesia. Lancet 2(7112):1123–1125
Small TN et al (1999) Association of reticular dysgenesis (thymic alymphoplasia and congenital aleukocytosis) with bilateral sensorineural deafness. J Pediatr 135(3):387–389
Bujan W et al (1993) Effect of recombinant human granulocyte colony-stimulating factor in reticular dysgenesis. Blood 82(5):1684
Pannicke U et al (2009) Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet 41(1):101–105
Lagresle-Peyrou C et al (2009) Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet 41(1):106–111
Lee HJ et al (2007) AK2 activates a novel apoptotic pathway through formation of a complex with FADD and caspase-10. Nat Cell Biol 9(11):1303–1310
Chediak MM (1952) New leukocyte anomaly of constitutional and familial character. Rev Hematol 7(3):362–367
Higashi O (1954) Congenital gigantism of peroxidase granules; the first case ever reported of qualitative abnormity of peroxidase. Tohoku J Exp Med 59(3):315–332
Menasche G et al (2000) Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 25(2):173–176
Jung J et al (2006) Identification of a homozygous deletion in the AP3B1 gene causing Hermansky-Pudlak syndrome, type 2. Blood 108(1):362–369
Bohn G et al (2007) A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med 13(1):38–45
Dell'Angelica EC (2007) Bad signals jam organelle traffic. Nat Med 13(1):31–32
Wei ML (2006) Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res 19(1):19–42
Clark RH et al (2003) Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat Immunol 4(11):1111–1120
Huizing M et al (2002) Nonsense mutations in ADTB3A cause complete deficiency of the beta3A subunit of adaptor complex-3 and severe Hermansky-Pudlak syndrome type 2. Pediatr Res 51(2):150–158
Shotelersuk V et al (2000) A new variant of Hermansky-Pudlak syndrome due to mutations in a gene responsible for vesicle formation. Am J Med 108(5):423–427
Kotzot D, Richter K, Gierth-Fiebig K (1994) Oculocutaneous albinism, immunodeficiency, hematological disorders, and minor anomalies: a new autosomal recessive syndrome? Am J Med Genet 50(3):224–227
Enders A et al (2006) Lethal hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type II. Blood 108(1):81–87
Dell'Angelica EC et al (1999) Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell 3(1):11–21
Sugita M et al (2002) Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity 16(5):697–706
Fontana S et al (2006) Innate immunity defects in Hermansky-Pudlak type 2 syndrome. Blood 107(12):4857–4864
Bonifacino JS, Dell'Angelica EC (1999) Molecular bases for the recognition of tyrosine-based sorting signals. J Cell Biol 145(5):923–926
Wunderlich W et al (2001) A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold mp1 on a late endosomal/lysosomal compartment. J Cell Biol 152(4):765–776
Kurzbauer R et al (2004) Crystal structure of the p14/MP1 scaffolding complex: how a twin couple attaches mitogen-activated protein kinase signaling to late endosomes. Proc Natl Acad Sci U S A 101(30):10984–10989
Teis D, Wunderlich W, Huber LA (2002) Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev Cell 3(6):803–814
Teis D et al (2006) p14-MP1-MEK1 signaling regulates endosomal traffic and cellular proliferation during tissue homeostasis. J Cell Biol 175(6):861–868
Melis D et al (2005) Genotype/phenotype correlation in glycogen storage disease type 1b: a multicentre study and review of the literature. Eur J Pediatr 164(8):501–508
Boztug K et al (2009) A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med 360(1):32–43
Bouatia-Naji N et al (2008) A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science 320(5879):1085–1088
Chen WM et al (2008) Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J Clin Invest 118(7):2620–2628
Zuelzer WW (1964) Myelokathexis"—a new form of chronic granulocytopenia. Report of a case. N Engl J Med 270:699–704
Gorlin RJ et al (2000) WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet 91(5):368–376
Hernandez PA et al (2003) Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet 34(1):70–74
Sanmun D et al (2006) Stromal-derived factor-1 abolishes constitutive apoptosis of WHIM syndrome neutrophils harbouring a truncating CXCR4 mutation. Br J Haematol 134(6):640–644
Kawai T et al (2007) WHIM syndrome myelokathexis reproduced in the NOD/SCID mouse xenotransplant model engrafted with healthy human stem cells transduced with C-terminus-truncated CXCR4. Blood 109(1):78–84
Balabanian K et al (2005) WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood 105(6):2449–2457
Kostmann R (1956) Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria. Acta Paediatr Suppl 45(Suppl 105):1–78
Nagata S et al (1986) Molecular cloning and expression of cDNA for human granulocyte colony-stimulating factor. Nature 319(6052):415–418
Skokowa J et al (2009) NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nat Med 15(2):151–158
Rosenberg PS et al (2006) The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood 107(12):4628–4635
Donini M et al (2007) G-CSF treatment of severe congenital neutropenia reverses neutropenia but does not correct the underlying functional deficiency of the neutrophil in defending against microorganisms. Blood 109(11):4716–4723
Germeshausen M, Ballmaier M, Welte K (2007) Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: results of a long-term survey. Blood 109(1):93–99
Germeshausen M, Welte K, Ballmaier M (2009) In vivo expansion of cells expressing acquired CSF3R mutations in patients with severe congenital neutropenia. Blood 113(3):668–670
Liu F et al (2008) Csf3r mutations in mice confer a strong clonal HSC advantage via activation of Stat5. J Clin Invest 118(3):946–955
Acknowledgements
We thank all clinical and scientific colleagues referring patients, Dr. Georg Bohn for providing a first draft, Birthe Landes for editorial help. This work was supported by the German José Carreras Foundation, BMBF networks on Congenital bone marrow failure syndromes and Primary immunodeficiency syndromes, and the Clotten Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Klein, C., Welte, K. Genetic Insights into Congenital Neutropenia. Clinic Rev Allerg Immunol 38, 68–74 (2010). https://doi.org/10.1007/s12016-009-8130-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-009-8130-5