
Abstract
Molecular dynamics (MD) simulations of wild-type and V91W mutant Mycobacterium tuberculosis-LprG (Mtb-LprG) were performed with the goal to provide a comprehensive understanding of the Mtb-LprG as a potential antimycobacterial target. A long-range MD simulations and post-MD analyzes led us to various results that plainly explained the impact of V91W mutation on Mtb-LprG. Herein, the results revealed that the wild-type is less stable compared to V91W mutant. This was further supported by root mean square fluctuation, where the V91W mutant showed a higher degree of flexibility compared to the wild-type. Dynamic cross-correlation analysis revealed that induced mutation leads to higher residual flexibility in the mutant structure as compared to the wild-type structure thus resulting in the existence of negatively correlated motions. The difference in principal component analysis scatter plot across the first two normal modes suggests a greater mobility of the V91W mutant conformation compared to the wild-type. Thermodynamic calculations revealed that the van der Waals (Evdw) forces contribute the most towards binding free energy in a case of the V91W mutant as compared to the wild-type LprG complex. In addition, the residue interaction networks revealed more of Evdw interaction existence among residues in case of the V91W mutant. This study supports the Mtb-LprG as a potential antimycobacterial target and also serves as a cornerstone to identifying new potential targets that have no inhibitors.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tuberculosis (TB) is one of the major causes of death in the world with 8.7 million new cases and 1.4 million deaths reported annually [1, 2]. Mycobacterium tuberculosis (Mtb), the etiological agent of this disease, has been estimated to infect one-third of world’s population and 1.5 million people die from its infection worldwide [2,3,4]. However, TB can be treated with an uninterrupted, multi-drug regimen of rifampicin (RIF), isoniazid (INH), pyrazinamide (PZA), and ethambutol (EMB) taken for 2 months followed by RIF and INH for 4 months [5,6,7]. The currently-used drugs mainly inhibit protein synthesis, mycolic acid biosynthesis, arabinogalactan biosynthesis, translation and trans-translation, mycolic acid synthesis, transcription, folate biosynthesis, DNA supercoiling and peptidoglycan synthesis [8,9,10]. Failure to complete the full course of treatment have led to drug-resistant Mtb with an estimation of 480,000 people worldwide developing multidrug-resistant TB (MDR-TB) in 2013 [11]. Drug resistance of Mtb against almost all currently-approved anti-TB drugs has motivated the urgent need for new effective drugs and drug targets for the advancement of drug discovery against Mtb.
Mtb has the capacity to synthesize and store large quantities of triacylglycerols (TAG) and during starvation, it catabolizes TAG as an energy source [4, 12, 13]. In addition, low immune function results in enhanced capacity to Mtb growth, especially in individuals infected with HIV, hence TB is considered number one of the causes of death in HIV/AIDS patients [14, 15]. The mycobacterial glycolipids, which are structural components of the cell wall, contribute to mycobacterial resistance to bactericidal free-radicals [16, 17]. Hence, Mtb is capable of long-term survival in the host during the periods of reduced growth and has the capacity to regrow rapidly [4].
LprG (Fig. 1) is a lipoprotein that plays a major role in transporting TAG from the cytoplasm to the outer membrane [4, 18, 19]. In addition, LprG is considered as one of the several lipoproteins responsible for optimal growth of Mtb in the host [20]. A large central cavity of LprG revealed to accommodate triacylated lipid species [4, 17]. Hence, native LprGs are able to transfer lipids at a high rate and yields as compared to mutant LprG-V91W, located within the hydrophobic cavity [4]. It is suggested that the cell wall components may contribute in several aspects of tuberculosis pathogenesis and virulence [2].

The front view of LprG (PDB code: 4ZRA) of the Mtb in complex with TAG: The Mtb-LprG chain contain Helix (red), Sheet (yellow) and Loop (green). TAG represented as blue dot ball
Recent work by Drage et al. [21], suggest that the pocket of LprG lipoprotein binds triacylated glycolipids, and introduction of a single mutation (valine 91 replaced with a tryptophan) in the binding pocket disrupt the glycolipid binding function of LprG, suggesting LprG as a potential target. Hence, the LprG (wild-type and V91W mutant) mechanism is not well established. Therefore, the current study provides molecular understanding of the lipolytic activity of LprG as a potential target also give a lead to the development of potent TB drugs, from a computational perspective.
In recent years, molecular dynamics (MD) simulation of protein molecules have been adopted to provide comprehensive understanding of the dynamic characteristics of proteins [22,23,24]. MD simulations have become the close counterpart to experiment in the understanding of complex systems at the atomic level [22, 24, 25]. In one of our recent papers, numerous post-dynamics analysis approaches, including binding free energy calculations, root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), principal component analysis (PCA) and dynamic cross correlation have proven to be useful approaches in understanding protein molecules [24].
In this work, we aim to provide a comprehensive understanding of the impact of the V91W mutation on the activity of Mtb-LprG in the transportation of TAG and provide insight into the future development of innovative chemotherapeutics against Mtb.
Due to computational efficiency regarding the LprG mechanism prompted us to perform a comprehensive analysis using MD approach. Post-dynamics analyzes were employed to understand the impact of LprG V91W mutation on upon TAG binding. To the best of our knowledge, this is the first account where such comprehensive computational tools are applied to reveal the impact of LprG V91W mutant upon TAG binding.
Findings reported in this study could aid in the understanding of the binding mechanism of TAG to LprG and LprG binding landscape, which in turn could pave way in the design new potential drugs.
Computational Methods
System Preparation
The X-ray crystal structure of LprG in complex with Tripalmitoylglycerol and TGA (PDB code: 4ZRA) [4] was used as the starting coordinates. Co-crystalized solvent molecules and ligand (TGA) were deleted during the preparation of the receptor structure. Hydrogen atoms were added to the isolated ligand. Mutation was carried out manually (Fig. 2) at position 91 to mutate Valine (V) into Tryptophan (W) using Chimera software.

Crystal structure of Mtb-LprG highlighting the position of the V91W mutation a and a wild-type b
Chimera software package [26] was used for structure preparation as well as residue mutation. The wild-type and V91W mutant systems were subjected to molecular dynamic simulations as described in the section 2.2.
Molecular Dynamic Simulations
MD simulations for wild-type and V91W mutant LprG in complex with TGA were performed using the GPU version of the PMEMD engine provided with the Amber14 software package [27]. To optimize the systems, ANTECHAMBER and LEaP module of Amber14 were used to ensure all parameters are present for MD simulations. The protein system parameters were determined with the FF12SB [28] variant of the Amber force field. The LEaP module of Amber14 was used for the addition of missing hydrogen atoms to the protein and counter ions addition to neutralize the systems. The systems were suspended within an orthorhombic box of TIP3P [29] water box such that all protein atoms were within 10 Å of a box edge. Long-range electrostatic interactions were treated with the Ewald method [30], a component of Amber14, with set parameters of direct space and a van der Waals cut-off of 12 Å. Prior to system preparations, the minimizations, heating and equilibration steps were performed as previously described in our recent report and a production MD run for continuous 400 ns was performed [31,32,33].
The trajectory in both system simulations were then saved and analyzed in every 1 ps. Post-MD analysis such as RMSD, RMSF, Radius of Gyration, dynamic cross correlation and PCA were carried out using the CPPTRAJ and PTRAJ modules [34, 35] of the Amber14 suite. Chimera molecular modeling tool and Origin data analysis software version 6 (http://www.originlab.com/) were carried out for all visualizations and plots, respectively [36].
Since Amber Tools has a tendency to re-number amino acids in a protein structure to a format recognized by Amber, Fig. 3 presents information on the amino acid sequence of 4ZRA crystal structure before and after MD simulation for clarity on amino acid numbering of analyzed structures.

RMSD for V91W mutant and Wild-type over 400 ns of simulation
Thermodynamic Calculations
Molecular Mechanics/Poisson–Boltzmann surface area (MM/PB-SA) is among the most popular approaches to study macromolecular stability and to estimate protein–ligand binding affinities [37,38,39]. MM/PBSA is more computationally efficient, hence, it can serve as a powerful tool in drug design. The binding free energy profiles of the TGA bound V91W mutant and wild-type variants of Mtb-LprG lipoprotein were computed using the Molecular Mechanics/Poisson–Boltzmann surface area (MM/PB-SA) approach. From each production run, the binding free energy was averaged over 1000 snapshots extracted from the 400 ns trajectory. The following set of equations describes the estimation of the binding free energy (ΔG):
where Egas signifies gas-phase energy; Eint signifies internal energy; Eele and Evdw represents the electrostatic and van der Waals contributions, respectively. The Egas was directly evaluated from the FF12SB force field terms. The solvation energy (Gsol) is the summation of contributions from the polar states, GGB, and non-polar states, GSA. The GGB is derived from solving the GB equation, whereas GSA contribution is estimated from the solvent accessible surface area (SASA) determined using a water probe radius of 1.4 Å. T and S represented the temperature and total solute entropy, respectively.
In order to obtain the contribution of each residue towards total binding free energy profile between the TGA and LprG, wild-type and V91W mutant, per-residue free energy decomposition analysis was computed at the atomic level using the MM/PBSA method in Amber 14. All ligand–protein interactions were performed using LigPlot [40].
Principal Component Analysis (PCA)
In this study, PCA was applied to give insight into the larger scale motions from individual MD trajectories and isolate the dominant modes of internal motion. After solvent and ions were first stripped off, PCA was performed on 400 ns MD trajectories using the PTRAJ and CPPTRAJ modules of Amber14. PCA analysis was performed on C-α atoms with 1000 snapshots extracted from the 400 ns trajectory. The first two principal components (PC1 and PC2) generated from trajectories were averaged for both wild-type and V91W mutant. Origin data analyzes program [36] was used to create the PCA scatter plot demonstrating the dominant conformational motion representative of each structure.
Dynamic Cross Correlation Matrices (DCCM)
In this study, dynamic cross correlation was calculated using the CPPTRAJ module incorporated in Amber14 to study the correlated motions of residual-based fluctuations during a 400 ns MD simulation. The equation below describes cross-correlation coefficient Cij for the pair of each C-α atoms i and j.
where Δrj and Δri is the displacement vectors correspond to jth and ith atom respectively. The cross-correlation coefficient Cij varies within a range of −1 to +1 of which the upper and lower limits correspond to a fully correlated and anti-correlated motion during the simulation process.
Analysis of Residue Interaction Networks (RINs)
RIN, a modern topology based analysis, assists in recognition of residue–residue contact difference in biological systems. The average structure derived from trajectory of each system, wild-type and V91W mutant, were used to construct the RINs interactively in 2D graphs using RING [41]. The RINs used in this work were defined using PROBE [42] software to identify interactions between residues in the proteins by evaluating their atomic packing. PROBE uses a small virtual probe (typically 0.25 Å) that is rolled around the van der Waals surface of each atom, and an interaction (contact dot) is detected if the probe touches another non-covalently bonded atom. Once interaction amino acids have been determined, RING uses several tools to define non-covalent interactions between amino acids (e.g., hydrogen bonds, salt bridges, pi–pi interaction, interatomic contact etc.).
Interactive visual analysis of residue networks
From MD averaged structures, RINs were generated and used to visualize the network using RINanalyzer [43] plugin integrated with Cytoscape [44]. In a RIN, the standard method described by Piovesan et al. [43] was adopted to analyze the nodes (which represent the protein amino acid residues) and the edges between them (which represent the type of interactions).
Results and Discussion
MD Simulations and Systems Stability
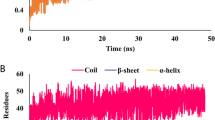
The RMSD was monitored to ensure that the systems were well equilibrated before further MD analysis. RMSD plot of simulated systems are provided in Fig. 3. It was noticed that the wild-type system was well stabilized throughout the simulation and the V91W mutant was well stabilized after a 100 ns time period.
During a 400 ns simulation, it can be observed that both systems stabilized, although, fluctuations in rigidity did increase during the 50–100 and 325–375 ns time period in the V91W mutant and wild-type, respectively. One possible explanation of this phenomenon is that the mutation induced changes in the flexibility of the lipoproteins, suggesting that the mutation affect the function of the lipoprotein. To gain more specific insight into the structural changes, we performed RMSF change between the two complexes.
Root Mean Square Fluctuation (RMSF)
The root mean square fluctuation (RMSF) provides insight into the flexibility of the protein structure regions [45]. In order to determine the amino acid flexibility for both Mtb-LprG V91W mutant and wild-type, RMSF of the protein backbone was calculated using Amber14 suite and presented in Fig. 4.

RMS fluctuations for the Mtb-LprG, V91W mutant and wild-type complex over 400 ns of simulation
In the present study, a conformational flexibility with a similar trend in fluctuations was observed in V91W mutant and wild-type. In Fig. 4, the wild-type has higher degree of flexibility compared to the V91W mutant.
The most significant changes can be seen on the amino acid residues for the following regions Ile145 and Pro148 located at the active site, showing higher fluctuation in case of the wild-type. However, the V91W mutant displayed lower fluctuation, for similar amino acid residues.
The results suggest that during the process of transporting TAG to the outer membrane, wild-type is more flexible compared to V91W mutant. Hence, as a result wild-type will be able to transfer lipids at a high rate and yield as compared to the V91W mutant. We can conclude from the results that induced mutation located at the active site leads to a conformational rigidity. In addition, this work support the experimental finding which suggested that the Mtb-LprG is able to transfer lipids at a high rate and yield as compared to V91W mutant [4].
Radius of Gyration
In this study, we computed the radius of gyration to give insight into the compactness of protein structures, providing insight into complex changes in the molecular shape during the 400 ns MD simulation [46,47,48]. Radius of gyration (Rg) plot of simulated systems are provided in Fig. 5, for V91W mutant and wild-type.

Radius of gyration comparison across the 400 ns MD simulation of V91W mutant and wild-type systems
Throughout the simulation (Fig. 5), the wild-type Mtb-LprG showed a similar Rg as compared to V91W mutant complex. Hence, both systems V91W mutant and wild-type share a similar Rg profile, which suggests that both have a similar degree of structural compactness.
Although both systems share a close resemblance, the wild-type system showed a slight increase in its Rg over time. The calculated Rg highly correlates with the estimated RMSF, which justified an increased biomolecular flexibility of wild-type structure as compared to V91W mutant structure. This results suggests that the Mtb-LprG appeared to be affected by the mutation at position 91.
Dynamic Cross-Correlation Analysis
The different correlation motions of both V91W mutant and wild-type system are plotted in Fig. 6. The plots are presented in different colors; strong correlated movements of specific residues were associated with highly positive regions range from yellow to red, while strong anti-correlated movements of specific residues associated with highly negative regions range from blue to black.

Dynamic cross-correlation matrix analyzes during 400 ns simulation for the a V91W mutant and b wild-type
The correlation map in Fig. 6 shows highly positive regions in case of wild-type as compared to V91W mutant. Hence, highly positive correlated residual motions in wild-type as compared to the V91W mutant occur between residue 20–20, 25–100, and 120–185 relative to each other. From the plot, the wild-type shows strong correlated residual motions, while the V91W mutant shows an existence of negatively correlated motions during 400 ns simulation time period.
From the plots, we can deduce that induced mutation at position 91 leads to an existence of negatively correlated motions. This observation correlates with that of RMSF and Rg, which justified that, the V91W mutant exhibits relatively higher biomolecular flexibility compared to wild-type lipoprotein. Hence, induced mutation located at the active site at position 91 leads to high RMSF and Rg consequently leading to an existence of negatively correlated motions.
Thermodynamic Calculations
Here we perform thermodynamic calculations to gain insight into the binding free energy profiles of Mtb-LprG binding to TAG. The relative binding free energy and the various energy components contribution of the protein–ligand complexes were calculated using the MM/PBSA approach. Table 1 shows the binding profiles of TAG bound with the V91W mutant and wild-type.
During MD simulation, the calculated binding free energy (∆Gbind) between TAG and V91W mutant is −104.675 and −99.961 kcal mol−1 in the case of wild-type. Hence, the results suggest that the ΔEvdw (−104.776 and −100.931 kcal mol−1) and ΔGgas (−125.390 and −107.159 kcal mol−1) contributions towards the total binding free energy in the TAG bound V91W mutant are higher than that for the TGA bound wild-type LprG complex, respectively.
Per-Residue Interaction Energy Decomposition Analysis
To estimate protein–ligand binding affinities and gain information on the important residues for ligand–protein interactions, molecular mechanics with generalized born and surface-area solvation (MM/GBSA) [49] approach was applied in this study. The total binding free energies for TAG was further decomposed into each Mtb-LprG amino acid residues contribution using the MM-GBSA method to understand ligand binding at an atomic level. Figure 7 shows the interacting amino acid residues with the ligand and per-residue energy decomposition analysis are shown in Fig. 8.

Representative structures for the LprG-TAG complexes: V91W mutant a and wild-type b, with graphical representation of the different binding forces

The V91W mutant a and wild-type b per residue graphs showing FBE contribution for TAG
As evident in Fig. 8, it can be observed from the energy decomposition analysis that the larger residual energy contributions were from LEU 36, LEU 41, ILE 94, and TYR 95 towards TAG binding to Mtb-LprG (V91W mutant and wild-type). The larger residual energy contributions (|ΔGbinding| > −3 kcal mol−1) in case of the V91W mutant were from LEU 36, ILE 94, and TYR 95, respectively, while LEU 41 (|ΔGbinding| < −2 kcal mol−1) shows less contribution as compared to other residues. On the other hand, residues TYR 95, LEU 36, and ILE 94, respectively, also show some major contributions towards the interaction with ΔEvdw of > −3 kcal mol−1.
In case of the wild-type, four residues (ILE 94, TYR 95, LEU 36, and LEU 41) had a major contribution (|ΔGbinding| > −2 kcal mol−1) to the total binding energy. LEU 36, ILE 94, and TYR 95 had a contribution of ΔEvdw > −2 kcal mol−1 in case of the wild-type.
We believe that this report provides invaluable information about the structural, dynamic and mechanistic features of Mtb-LprG as a potential drug target. Considering computational efficiency, energy-based pharmacophore [50] map approach can serve as a powerful tool in rational drug design of novel against Mtb, targeting LprG as a new target.
Principal Component Analysis (PCA)
The conformational motions of two systems were projected along the first two principal components (PC1 vs PC2) in order to gain further understanding of V91W mutant and wild-type conformation of Mtb-LprG. PCA was conducted taking into account the C-alpha atoms of residues of both systems. Figure 9 highlights the dominant changes in motion across principal components in the case of V91W mutant and wild-type configurations of Mtb-LprG. Amino acid fluctuation for both Mtb-LprG V91W mutant and wild-type, was calculated across principal components (PC1 and PC2) and presented in Fig. 10.

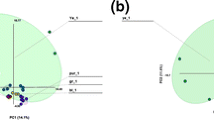
Projections of Eigen values during simulation period for a V91W mutant and b wild-type conformations of Mtb-LprG along the first two principal components (PC1 and PC2)

Residue-wise loading for PC1 (black) and PC2 (blue) for the Mtb-LprG V91W mutant a and wild-type b complex over 400 ns of simulation
To understand the mutational effect on the macromolecular conformation we utilized ProDy plugin integrated with VMD to generate porcupine plots corresponding to first two normal modes in each case [51,52,53]. The color scale from red to blue depicts high to low atomic displacements.
From the scatter plot it was observed that the wild-type complex occupies a larger phase space as compared to the V91W mutant complex. As evident in Fig. 9, wild-type complex residues exhibits a higher fluctuation as compared to the V91W mutant complex. The results plotted in Fig. 10 for V91W mutant and wild-type in the present study shows to follow similar trends to those reported for RMSF. From the plots, fluctuations >0.5 Å in case of the wild-type as compared to the V91W mutant with <0.3 Å were observed.
The results from the porcupine plots are in agreement with the PCA scatter plot and residue based fluctuation plots across different modes. These results provide solid information in an attempt to understand the dynamic behavior of Mtb-LprG Fig. 11.

Porcupine plots for the first two normal modes showing motion difference among variants of Mtb-LprG complexes. a, b Corresponds to V91W mutant and wild-type, respectively
Residue Interaction Network (RIN)
The network analysis of the protein backbone is a one of the strategy used to identify key residue interactions and can be used to explore the difference in RINs between different proteins including V91W mutant and wild-type [24, 32, 33]. In this work, we investigated the topology based interaction difference among key residues by generating RINs using the representative average structures from the MD simulation. Figure 12 highlights the RIN plots.

Comparison of residue interaction networks (RINs) of the average MD structures between the a V91W mutant and b wild-type variant of Mtb-LprG, highlighting changes in network interaction at point 91
From the RIN profile of the average MD structures, 289 edges were created in case of the mutation as compared to the wild type with 290 edges. As evident from the RIN plots (Fig. 12), it is clear that the presence of V91W mutation distorted the overall RIN when compared to wild type. In an interesting manner, there is a hydrogen bond and van der Waals force interaction between VAL56 (VAL91) and ALA67 (ALA102) whereas in the case of the mutant where VAL51 has been mutated to TRP56 (TRP91), only hydrogen bond interaction between TRP56 (TRP91) and ALA67 (ALA102) was observed. In addition, van der Waals force interaction between VAL56 (VAL91) and LEU38 (73) was observed in case of the wild type whereas in the case of the mutant TRP56 form a van der Waals force interaction with LEU38 (LEU73) and LEU41 (LEU76). Hence, these results are correlated to those of thermodynamic calculation which justified that the calculated van der Waals (−104.776 and −100.931 kcal mol−1) contributions more in case of the V91W mutant as compared to the wild-type LprG complex, respectively. The V91W mutation affects the interaction network, which ultimately affects the protein backbone and consequently the ligand binding landscape.
Conclusion
Drug-resistant Mtb against almost all currently approved anti-TB drugs has motivated the urgent need for new effective drugs and new drug targets for the advancement of drug discovery against Mtb. In this report, we embarked on various computational approaches in order to provide information on Mtb-LprG as a new drug target. MD simulations and post-MD analyzes led us to several findings that clearly explained the impact of V91W mutation on Mtb-LprG. RMSF, Rg and PCA analyzes for the wild-type system suggested a more flexible conformational nature of Mtb-LprG compared to the V91W mutant. On the other hand, DCC results shows to correlate with those of RMSF and Rg, which justified that the wild-type exhibit greater biomolecular flexibility. The induced mutation results in reduced residual flexibility, revealed by lower RMSF and Rg, leading to an existence of negative correlation motions. The study identified the V91W mutant with higher binding affinities as wild-type with the majority of the favorable binding energy contributions arise from ∆Evdw. Hence, thermodynamics calculations are in agreement with RIN analysis of the protein backbone which suggested more of van der Waals force interactions in case of the V91W mutant as compared to the wild-type. LprG represent novel candidate not targeted by existing TB drugs, and the availability of comprehensive understanding of the Mtb-LprG activity in the transportation of TAG greatly opens new opportunities for the development of new antibiotics to fight Mtb. This study not only suggest Mtb-LprG as a potential target, but also provide more insight into the structural, and dynamic mechanistic features of Mtb-LprG. Hence, the approach can also serve as a cornerstone to identifying new potential targets that have no inhibitors.
References
Naidoo, A., Naidoo, K., Yende-zuma, N., & Gengiah, T. N. (2015). Changes to antiretroviral drug regimens during integrated TB-HIV treatment: Results of the SAPiT trial. NIH Public Access, 19(2), 161–169. https://doi.org/10.3851/IMP2701.Changes.
Gaur, R. L., Ren, K., Blumenthal, A., Bhamidi, S., Gibbs, S., Jackson, M. et al. (2014). LprG-mediated surface expression of lipoarabinomannan is essential for virulence of Mycobacterium tuberculosis. PLoS Pathogens. https://doi.org/10.1371/journal.ppat.1004376.
Who. (2015). Global tuberculois report 2015—executive summary. World Health Organisation. https://doi.org/10.1037/e530172011-002.
Martinot, A. J., Farrow, M., Bai, L., Layre, E., Cheng, T.-Y., Tsai, J. H., & Rubin, E. J. (2016). Mycobacterial metabolic syndrome: LprG and Rv1410 regulate triacylglyceride levels, growth rate and virulence in Mycobacterium tuberculosis. PLOS Pathogens, 12(1), e1005351 https://doi.org/10.1371/journal.ppat.1005351.
Jnawali, H. N., & Ryoo, S. (2013). First and second line drugs and drug resistance. Tuberculosis-Current Issues in Diagnosis and Management, 163–180. https://doi.org/10.5772/54960
Poce, G., Cocozza, M., Consalvi, S., & Biava, M. (2014). SAR analysis of new anti-TB drugs currently in pre-clinical and clinical development. European Journal of Medicinal Chemistry, 86(April), 335–351. https://doi.org/10.1021/jm500131s.
Palomino, J., & Martin, A. (2014). Drug resistance mechanisms in Mycobacterium tuberculosis. Antibiotics, 3(3), 317–340. https://doi.org/10.3390/antibiotics3030317.
Shi Wanliang, Z. X. (2012). Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis: a potenatial mechnism for shortening the duration of chemotrerapy. Science, 333(6049), 1630–1632. https://doi.org/10.1126/science.1208813.Pyrazinamide.
Chakraborty, S., Gruber, T., Barry, C. E., Boshoff, H. I., & Rhee, K. Y. (2013). Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science, 339(6115), 88–91. https://doi.org/10.1126/science.1228980.
Zumla, A., Nahid, P., & Cole, S. T. (2013). Advances in the development of new tuberculosis drugs and treatment regimens. Nature Reviews Drug discovery, 12(5), 388–404. https://doi.org/10.1038/nrd4001.
WHO What is multidrug-resistant tuberculosis and how do we control it? (n.d.). http://www.who.int/features/qa/79/en/.
Alvarez, H. M., & Steinbüchel, a (2003). Triacylglycerols in prokaryotic microorganisms. Applied Microbiology and Biotechnology, 60(4), 367–376. https://doi.org/10.1007/s00253-002-1135-0.
Deb, C., Daniel, J., Sirakova, T. D., Abomoelak, B., Dubey, V. S., & Kolattukudy, P. E. (2006). A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. Journal of Biological Chemistry, 281(7), 3866–3875. https://doi.org/10.1074/jbc.M505556200.
Ngowi, B. J., Mfinanga, S. G., Bruun, J. N., & Morkve, O. (2008). Pulmonary tuberculosis among people living with HIV/AIDS attending care and treatment in rural northern Tanzania. BMC Public Health, 8, 341 https://doi.org/10.1186/1471-2458-8-341.
Trinh, Q. M., Nguyen, H. L., Nguyen, V. N., Nguyen, T. Va, Sintchenko, V., & Marais, B. J. (2015). Tuberculosis and HIV co-infection-focus on the Asia-Pacific region. International Journal of Infectious Diseases, 32, 170–178. https://doi.org/10.1016/j.ijid.2014.11.023.
Chan, J., Fujiwara, T., Brennan, P., McNeil, M., Turco, S. J., Sibille, J. C., & Bloom, B. R. (1989). Microbial glycolipids: possible virulence factors that scavenge oxygen radicals. Proceedings of the National Academy of Sciences of the United States of America, 86(7), 2453–2457. https://doi.org/10.1073/pnas.86.7.2453.
Shukla, S., Rojas, R. E., Seshadri, C., & Moody, D. B. (2011). NIH public access. Nature Structural Biology, 17(9), 1088–1095. https://doi.org/10.1038/nsmb.1869.Mycobacterium.
Ortalo-Magné, A., Lemassu, A., Lanéelle, M. A., Bardou, F., Silve, G., Gounon, P., & Daffé, M. (1996). Identification of the surface-exposed lipids on the cell envelopes of Mycobacterium tuberculosis and other mycobacterial species. Journal of Bacteriology, 178(2), 456–461.
Daffé, M., & Draper, P. (1997). The envelope layers of mycobacteria with reference to their pathogenicity. Advances in Microbial Physiology. https://doi.org/10.1016/S0065-2911(08)60016-8.
Rezwan, M., Grau, T., Tschumi, A., & Sander, P. (2007). Lipoprotein synthesis in mycobacteria. Microbiology, 153(3), 652–658. https://doi.org/10.1099/mic.0.2006/000216-0.
Drage, M. G., Tsai, H. C., Pecora, N. D., Cheng, T. Y., Arida, A. R., Shukla, S., & Harding, C. V. (2010). Mycobacterium tuberculosis lipoprotein LprG (Rv1411c) binds triacylated glycolipid agonists of Toll-like receptor 2. Nature Structural and Molecular Biology, 17(9), 1088–1095. https://doi.org/10.1038/nsmb.1869.
Adcock, S. A., & McCammon, J. A. 2006). Molecular dynamics: Survey of methods for simulating the activity of proteins. Chemical Reviews. https://doi.org/10.1021/cr040426m.
Hospital, A., Goñi, J. R., Orozco, M., & Gelpi, J. (2015). Molecular dynamics simulations: Advances and applications. Advances and Applications in Bioinformatics and Chemistry, 8, 37–47. https://doi.org/10.2147/AABC.S70333.
Moonsamy, S., Bhakat, S., Walker, R. C., & Soliman, M. E. (2016). Single active site mutation causes serious resistance of HIV reverse transcriptase to lamivudine: Insight from multiple molecular dynamics simulations. Cell Biochemistry and Biophysics, 74(1), 35–48. https://doi.org/10.1007/s12013-015-0709-2.
Asamitsu, K., Hirokawa, T., Hibi, Y., & Okamoto, T. (2015). Molecular dynamics simulation and experimental verification of the interaction between cyclin T1 and HIV-1 Tat proteins. PLoS One, 10(3), 1–17. https://doi.org/10.1371/journal.pone.0119451.
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational Chemistry, 25(13), 1605–12. https://doi.org/10.1002/jcc.20084.
Case, D. a., Cheatham, T. E., Darden, T., Gohlke, H., Luo, R., Merz, K. M., & Woods, R. J. (2005). The Amber biomolecular simulation programs. Journal of Computational Chemistry, 26(16), 1668–1688. https://doi.org/10.1002/jcc.20290.
Perez, A., MacCallum, J. L., Brini, E., Simmerling, C., & Dill, K. A. (2015). Grid-based backbone correction to the ff12SB protein force field for implicit-solvent simulations. Journal of Chemical Theory and Computation, 11(10), 4770–4779. https://doi.org/10.1021/acs.jctc.5b00662.
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics, 79(2), 926 https://doi.org/10.1063/1.445869.
Harvey, M. J., & De Fabritiis, G. (2009). An implementation of the smooth particle mesh Ewald method on GPU hardware. Journal of Chemical Theory and Computation, 5(9), 2371–2377. https://doi.org/10.1021/ct900275y.
Chetty, S., & Soliman, M. E. S. (2015). Possible allosteric binding site on Gyrase B, a key target for novel anti-TB drugs: Homology modelling and binding site identification using molecular dynamics simulation and binding free energy calculations. Medicinal Chemistry Research, 24(5), 2055–2074. https://doi.org/10.1007/s00044-014-1279-3.
Bhakat, S., Martin, A. J. M., & Soliman, M. E. S. (2014). An integrated molecular dynamics, principal component analysis and residue interaction network approach reveals the impact of M184V mutation on HIV reverse transcriptase resistance to lamivudine. Molecular BioSystems, 10(8), 2215–28. https://doi.org/10.1039/c4mb00253a.
Karubiu, W., Bhakat, S., & Soliman, M. E. S. (2014). Compensatory role of double mutation N348I/M184V on nevirapine binding landscape: Insight from molecular dynamics simulation. The Protein Journal. https://doi.org/10.1007/s10930-014-9576-8.
Roe, D. R., & Cheatham, T. E. (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9(7), 3084–3095. https://doi.org/10.1021/ct400341p.
Case, D., Berryman, J. T., Betz, R. M., Cerutti, D. S., Cheatham, T. E., Darden, T. et al. (2015). AMBER 2015. University of California, San Francisco.
Binder, J. (2007). The origin of graphing and analysis software. Aerospace America, 45(9), 20.
Zhou, Z., & Madura, J. D. (2004). Relative free energy of binding and binding mode calculations of HIV-1 RT inhibitors based on dock-MM-PB/GS. Proteins: Structure, Function and Genetics, 57(3), 493–503. https://doi.org/10.1002/prot.20223.
Zhigang, Z., Yanli, W., & Bryant, S. H. (2009). Computational analysis of the cathepsin B inhibitors activities through LR-MMPBSA binding affinity calculation based on docked complex. Journal of Computational Chemistry, 30(14), 2165–2175. https://doi.org/10.1002/jcc.21214.
Genheden, S., & Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opinion on Drug discovery, 10(5), 449–61. https://doi.org/10.1517/17460441.2015.1032936.
Laskowski, R. A., & Swindells, M. B. (2011). LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling, 51(10), 2778–2786. https://doi.org/10.1021/ci200227u.
Doncheva, N. T., Klein, K., Domingues, F. S., & Albrecht, M. (2011). Analyzing and visualizing residue networks of protein structures. Trends in Biochemical Sciences. https://doi.org/10.1016/j.tibs.2011.01.002.
Word, J. M., Lovell, S. C., LaBean, T. H., Taylor, H. C., Zalis, M. E., Presley, B. K., & Richardson, D. C. (1999). Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. Journal of Molecular Biology, 285(4), 1711–33. https://doi.org/10.1006/jmbi.1998.2400.
Piovesan, D., Minervini, G., & Tosatto, S. C. E. (2016). The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Research. https://doi.org/10.1093/nar/gkw315.
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., & Ideker, T. (2003). Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. https://doi.org/10.1101/gr.1239303.
Bornot, A., Etchebest, C., & De Brevern, A. G. (2011). Predicting protein flexibility through the prediction of local structures. Proteins: Structure, Function and Bioinformatics, 79(3), 839–852. https://doi.org/10.1002/prot.22922.
Pan, L., & Patterson, J. C. (2013). Molecular dynamics study of Zn(Aβ) and Zn(Aβ)2. PLoS One. https://doi.org/10.1371/journal.pone.0070681
Lobanov, M. I., Bogatyreva, N. S., & Galzitskaia, O. V. (2008). Radius of gyration is indicator of compactness of protein structure. Molekuliarnaia Biologiia, 42(4), 701–706. https://doi.org/10.1134/S0026893308040195.
Caves, L. S., Evanseck, J. D., & Karplus, M. (1998). Locally accessible conformations of proteins: multiple molecular dynamics simulations of crambin. Protein Science: A Publication of the Protein Society, 7(3), 649–66. https://doi.org/10.1002/pro.5560070314.
Godschalk, F., Genheden, S., Söderhjelm, P., & Ryde, U. (2013). Comparison of MM/GBSA calculations based on explicit and implicit solvent simulations. Physical Chemistry Chemical Physics, 15(20), 7731–9. https://doi.org/10.1039/c3cp00116d.
Kumalo, H. M., & Soliman, M. E. (2016). Per-residue energy footprints-based pharmacophore modeling as an enhanced in silico approach in drug discovery: A case study on the identification of novel??-Secretase1 (BACE1) inhibitors as anti-Alzheimer agents. Cellular and Molecular Bioengineering, 9(1), 175–189. https://doi.org/10.1007/s12195-015-0421-8.
Humphrey, W., Dalke, A., & Schulten, K. (1996). VMD: Visual molecular dynamics. Journal of Molecular Graphics, 14(1), 33–38. https://doi.org/10.1016/0263-7855(96)00018-5.
Bakan, A., Meireles, L. M., & Bahar, I. (2011). ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics, 27(11), 1575–1577. https://doi.org/10.1093/bioinformatics/btr168.
Bakan, A., Dutta, A., Mao, W., Liu, Y., Chennubhotla, C., Lezon, T. R., & Bahar, I. (2014). Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics, 30(18), 2681–2683. https://doi.org/10.1093/bioinformatics/btu336.
Acknowledgements
The authors acknowledge the School of Health Sciences, University of KwaZulu-Natal, Westville Campus, for financial support and the Center for High Performance Computing (CHPC, http://www.chpc.ac.za), Cape Town, Republic of South Africa, for high-performance computing resources.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Machaba, K.E., Mhlongo, N.N. & Soliman, M.E.S. Induced Mutation Proves a Potential Target for TB Therapy: A Molecular Dynamics Study on LprG. Cell Biochem Biophys 76, 345–356 (2018). https://doi.org/10.1007/s12013-018-0852-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-018-0852-7