
Abstract
Altered expressions of microRNAs (miRNAs) are reported in pancreatic cancer and associate with cancer pathogenesis, apoptosis, and cell growth, thereby functioning as either tumor suppressors or oncogenes. However, the majority of studies focus on defining the regulatory functions of miRNAs, whereas few investigations are directed toward assessing how the miRNA themselves are transcriptionally regulated. In this study, integration of published multi-level expression data and bioinformatics computational approach was used to predict two regulation mechanisms: transcription factors (TF)–miRNA–mRNA regulation and long non-coding RNA(lncRNA)–miRNA–mRNA regulation. To identify differentially expressed mRNAs, miRNAs, and lncRNAs, we integrated microarray expression data in pancreatic cancer tissues and normal tissues. Combination of differentially expressed mRNAs and miRNAs with miRNA–mRNA interactions based on crosslinking and immunoprecipitation followed by high-throughput sequencing (CLIP-Seq) data from StarBas, we constructed miRNA–mRNA regulatory network. Then we constructed two regulatory networks including TF–miRNA–mRNA and lncRNA–miRNA–mRNA based on chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-Seq) data from ChIPBase and CLIP-Seq data. A total of 4385 mRNAs, 500 miRNAs, and 21 lncRNAs were differentially expressed, of which, 18 mRNAs and 54 miRNAs are with high confidence. In miRNA–mRNA regulatory network, interrelated miRNAs target 1701 differentially regulated mRNAs. By constructing regulatory network, 19miRNAs including hsa-miR-137, hsa-miR-206, hsa-miR-429, hsa-miR-320d, and hsa-miR-320c are predicted to participate in lncRNA–miRNA–mRNA regulation. Furthermore, 8 miRNAs including hsa-mir-137, hsa-mir-206, hsa-mir-429, hsa-mir-375, hsa-mir-326, hsa-mir-217, hsa-mir-301b, and hsa-mir-184 are predicted to participate in TF–miRNA–mRNA regulation. In an integrated data analysis, we reveal large-scale effects of interrelated miRNAs and provide a model for predicting the mechanism of miRNAs disorder. Our study provides a new insight into understanding the transcriptional regulation of pancreatic cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The increasing availability of multi-level expression data from cancer and normal tissue has created a new opportunity for integration and extraction of knowledge from large datasets such as gene expression omnibus (GEO) that promises a more comprehensive understanding of cancer. Previous data integration efforts in pancreatic cancer have focused on integrating a subset of profiles. For example, Tahira et al. [19] used custom complementary DNA (cDNA) microarray comprising protein-coding messenger RNA (mRNA) and long noncoding RNA (lncRNA) to identify significant expression signatures correlated to pancreatic cancer and metastasis. In addition, Frampton et al. [7] combined data from microRNA (miRNA) and mRNA expression profiles and bioinformatic analyses to identify functional miRNA–mRNA interactions that contribute to growth of PDACs. Similarly, Donahue et al. [5] developed a method to identify prognosis-significant genes based on analysis of DNA copy number, and mRNA and miRNA expression. However, it remains unknown what is their relationships and how to efficiently integrate different level expression profiles.
Recent studies have revealed that miRNAs disorder is often due to the aberrant expression of lncRNAs and transcription factors (TFs). lncRNAs because of their longer size (>200 nucleotides), can regulate microRNA abundance by binding and sequestering them, acting as the so-called microRNA sponges, thus regulating the expression of target mRNAs. Wang et al. close the circle by describing a lncRNA–miRNA–mRNA (CHRF-miR489-Myd88) trio that functions interdependently to regulate cardiac hypertrophy. HULC is a highly up-regulated lncRNA in liver cancer transcribed from human chromosome 6p24.3. The HULC gene consists of two exons and a single intron, while HULC contains a polyA tail and particularly a conserved target site of miR-372 [16, 20]. The expression of miRNAs is controlled by TFs also. For example, miR-122 is transcriptionally controlled by TFs enriched in the liver, such as hepatocyte nuclear factors (HNFs) and CCAAT/enhancer-binding proteins (C/EBPs), which play pivotal roles in regulating the expression of liver-specific genes [22].
Recent advances in high-throughput sequencing of immunoprecipitated RNAs after crosslinking (CLIP-Seq, HITS-CLIP, PAR-CLIP, CLASH, and iCLIP) and chromatin immunoprecipitation with massively parallel DNA sequencing (ChIP-Seq) provide powerful ways to identify biologically relevant miRNA–mRNA and lncRNA–miRNA and TF–miRNA interactions [ 13, 23, 24 ]. The application of CLIP-Seq methods has reliably identified Argonaute (Ago) and other RNA-binding proteins (RBP) binding sites to characterize miRNA–mRNA and lncRNA–miRNA interactions [ 1, 3, 9, 12 ]. The application of the ChIP-Seq technique has significantly decreased false-positive predictions of transcription factor binding sites (TFBSs) to identify TF–miRNA interactions [6, 17, 18].
The involvement of plenty of lncRNAs and TFs in the transcriptional regulation of miRNA has not been reported in pancreatic cancer. There is a great need to integrate these large-scale data sets to explore the regulation mechanisms of TF–miRNA–mRNA and lncRNA–miRNA–mRNA. In this study, we primarily use bioinformatics method to predict these two regulation mechanism of miRNAs disorder in pancreatic cancer. We integrated expression microarray to identify a differentially expressed mRNAs, miRNAs, and lncRNAs in pancreatic cancer. Combining these differentially expressed RNAs, we constructed miRNA–mRNA regulatory network based on CLIP-Seq data to reveal biological effects of interrelated miRNAs. Furthermore, analysis of regulatory networks including TF–miRNA–mRNA and lncRNA–miRNA–mRNA identified several miRNAs, lncRNAs, and TF which are possibly involved in this two regulation mechanisms in pancreatic cancer.
Materials and Methods
Selection of Studies and Datasets
GEO (http://www.ncbi.nlm.nih.gov/geo/webcite) was searched for PADC mRNAs, miRNAs, and lncRNAs expression profiling studies. We included only original experimental articles that compared the expression of RNAs in PDAC tissue and noncancerous pancreatic tissue in humans. The titles and abstracts of the articles were screened, and the full text of the articles of interest was evaluated. We selected three datasets including GSE32676 (25 pancreatic cancer samples and seven normal pancreas control), GSE30134 (18 pancreatic cancer samples and nine normal pancreas control), and pancreatic expression database [4] (PED, http://www.pancreasexpression.org/webcite) (96 pancreatic cancer samples and four normal pancreas control) as original mRNAs datasets finally. GSE30134 is used as an original lncRNAs dataset. Another three datasets including GSE24279 (136 pancreatic cancer samples and 22 normal pancreas controls), GSE28862 (three pancreatic cancer samples and three normal pancreas adjacent to cancer), and GSE32678 (25 pancreatic cancer samples and seven normal pancreas controls) were selected as original miRNAs dataset. GSE32678 and GSE32676 were came from the same group of the patients.
Data Processing
Differential Expression Analysis
We downloaded the original data and documents from these datasets with CEL or TXT format. If the data included raw CEL data, we use GC-RMA methods from bioConductor (http://www.bioconductor.org/) to normalize and summarize the probe set information. For which had no CEL data, we used standard TXT format for import. Then the data file was imported into BRB-ArrayTools version 4.2 [ 25 ] (National Cancer Institute), which was available at http://linus.nci.nih.gov/BRB-ArrayTools.html. RNAs which passed filtering and normalization criteria were analyzed using BRB-ArrayTools, which compares RNAs expression among predefined classes and presumes the data consists of experiments of different samples representative of the classes. We identified differentially expressed RNAs using a multivariate permutation test.
Vote-Counting Strategy
The RNAs were selected according to their importance as follows: (i) number of the same RNAs being differentially expressed in expression profiling datasets; (ii) number of the same RNAs having a consistent direction of change; and (iii) RNAs with an inconsistent direction of change in only two datasets were excluded.
Construction of Regulatory Network
A total of 606408 miRNA–mRNA interactions and 10212 lncRNA–miRNA interactions were downloaded based on CLIP-Seq data from starBas [13, 23] (http://starbase.sysu.edu.cn/index.php/webcite) in April, 2014. Of these, 4811 pairs of miRNA-mRNA interactions have valid relationships with expression profiles, which were preprocessed and identified to differentially expressed miRNA and mRNA in pancreatic cancer. The following parameters were selected for reducing false positives in processing: (i) Number of supporting experiments ≥1 mean that at least one CLIP-Seq experiments supported the predicted miRNA target site. (ii) Pan-Cancer ≥1 mean that expression of miRNA and target gene is anti-correlation (pearson correlation: r < 0, p value < 0.05) at least one cancer type. (iii) Expression regulatory patterns of miRNA–mRNA, consistent with up-down or down-up were included. A total of 55675 TF–miRNA interactions were downloaded based on ChIP-Seq data from ChIPBase [24] (http://deepbase.sysu.edu.cn/chipbase/index.php/webcite) in April, 2014. The above interactions information was imported into Cytoscape software version 2.8.3 [2] (http://www.cytoscape.org/webcite) to construct regulatory network.
Pathway Analysis
To explore biological effects of interrelated miRNAs in pancreatic cancer, we inputted the candidate genes into database of DAVID [8] (http://david.abcc.ncifcrf.gov/webcite) for pathway enrichment analysis.
Results
Identification of Differentially Expressed mRNAs, miRNAs and lncRNAs
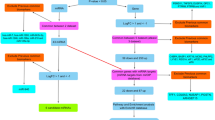
We collected pancreatic cancer-related microarray expression data including mRNAs, miRNAs, and lncRNAs from GEO:139 pancreatic cancers and 20 normal pancreas in three mRNAs expression data sets; 164 pancreatic cancers and 32 normal pancreas in three miRNAs expression datasets; and 18 pancreatic cancers and nine normal pancreas in one lncRNAs expression data sets. We developed a computational pipeline to analyze the above date (Fig. 1). The results show that 4385 mRNAs, 500 miRNAs, and 21 lncRNAs were differentially expressed in pancreatic cancer (Tables S1, S2, Table 1). Of which, 18 mRNAs and 54 miRNAs were of high confidence (Tables 2, 3). However, 325 mRNAs and 45 miRNAs with inconsistent direction in two studies were excluded (Table S3, Table S4).

Flow-chat of data analysis. Firstly, we identified differentially expressed mRNAs, miRNAs, and lncRNAs in pancreatic cancer through integrating expression microarray data Then we constructed network of miRNA–mRNA based on CLIP-Seq data. Furthermore, we constructed lncRNA–miRNA–mRNA regulatory network based on CLIP-Seq data and TF–miRNA–mRNA regulatory networks based on ChIP-Seq data from ChIPBase
Regulatory Network of lncRNA–miRNA–mRNA and IT–miRNA–mRNA
We construct a miRNA–mRNA regulatory network. Interaction analysis show that 36 differentially expressed miRNAs targeted 1779 mRNAs up or down. In detail, 18 down-expressed miRNAs deregulate 1170 mRNA and 18 over-expressed miRNAs deregulate 609 mRNAs (Fig. 2). As an typical example, interaction of miR217-KRAS has been found in this miRNA–mRNA regulatory network. miR-217 has been found to down-regulated in PDAC tissues and in PDAC cell lines compared with the corresponding normal pancreatic tissue. KRAS was proved to be a direct target of miR-217 by dual-luciferase reporter gene assay. Previous study showed that miR-217 can regulate KRAS and function as a tumor suppressor in PDAC [26]. The regulation of miR217-KRAS was found in this miRNA–mRNA regulatory network which validated our predicted results. miR-326 has been reported to down-regulated in glioblastoma specimens. PKM2 as target of miRNA-326 was high levels of protein expression [10]. The regulation of miR326-PKM2 now was found in pancreatic cancer, however, the role of miR-326 in pancreatic cancer has not been elucidated thus far. miR-125a has been reported to degradate SMG1 mRNA expression in human cells [21]. SMG1 is considered to be an essential factor in the nonsense-mediated mRNA decay pathway. This interaction of miR-125a-SMG1 identified in this study has not been investigated in pancreatic cancer.

The miRNA–mRNA regulatory network. miRNA was indicated to triangle and mRNA was indicated to circle. The color of red represents high expression and green represents low expression. Arrows indicated miRNAs regulating mRNA and straight line indicated miRNAs deregulating mRNA (Color figure online)
In lncRNA–miRNA–mRNA regulatory network, abnormal expression of 19 miRNAs was regulated by the aberrant expression of lncRNAs (Fig. 3). Additionally, abnormal eight miRNAs were transcriptionally regulated by TFs in IT–miRNA–mRNA regulatory network (Fig. 4). Three lncRNAs including MALAT1, HOTAIR, and H19 have been reported to participate in gene expression in pancreatic cancer. Expression of MALAT1 was significantly higher in PDAC compared to the adjacent normal pancreatic tissues and patients with higher MALAT1 expression had a poorer disease-free survival [14]. HOTAIR expression was increased in pancreatic tumors compared with non-tumor tissue and is associated with more aggressive tumors [11]. H19 was reported as a sponge to antagonize let-7 in pancreatic cancer [15]. The involvement of several lncRNAs and TFs in the transcriptional regulation of miRNA was rarely reported in pancreatic cancer.

The regulatory network of lncRNA–miRNA–mRNA. lncRNA, miRNA, and mRNA were indicated to deformed triangle, triangle, and circle, respectively. The color of red represents high expression and green represents low expression. Arrows indicated miRNAs regulating mRNA and straight line indicated miRNAs deregulating mRNA. And the purple line indicated lncRNA sponging miRNA (Color figure online)

The regulatory network of TF–miRNA–mRNA. TF, miRNA and mRNA were indicated to diamond, triangle, and circle, respectively. The color of red represents high expression and green represents low expression. Arrows indicated miRNAs regulating mRNA and straight line indicated miRNAs deregulating mRNA. And the azure color line indicated TF binding to miRNA (Color figure online)
Pathway Annotation of Differentially Expressed miRNAs
In order to study the function of differentially expressed miRNAs in regulatory network, we performed KEGG pathway annotation using DAVID Database for 21 miRNA in regulatory network and KEGG pathways of miRNA targeted genes are shown in Fig. 5. The most of differentially expressed miRNAs affected pathways included pathways in cancer; ECM-receptor interaction; focal adhesion; wnt signaling pathway; cell cycle; and TGF-beta signaling pathway.

Pathway annotation of 21 differential expressed miRNAs. Genes regulated by 21 differential expressed miRNAs were enriched in pathways using DAVID Database, respectively. Total 20 differential expressed miRNAs regulated genes have enriched significant pathways as shown. The area proportion of each pathway presents the number of genes enriched in this pathway
Discussion
In this study, we systematically analyze the complex effects of interrelated miRNAs and provide a framework for revealing the mechanism of miRNAs disorder regulated by TF–miRNA–mRNA and lncRNA–miRNA–mRNA. Conventional analysis methods focus on differentially expressed genes and miRNAs between biological processes or disease states, which then were selected for target prediction by bioinformatic analysis softwares such as TargetScan. Here, we integrated expression microarray to identify differentially expressed mRNAs and miRNAs. We especially combined public miRNAs–mRNAs interactions that have been generated by high-throughput CLIP-Seq to reduce the rate of false-positive predictions. Then we firstly constructed two regulatory networks including TF–miRNA–mRNA and lncRNA–miRNA–mRNA in pancreatic cancer. Our results revealed a set of miRNAs (Table 4) that were possibly involved in this two regulation mechanisms. This study provides a new insight into understanding the molecular mechanism of pancreatic cancer.
However, this study had some limitations that should be acknowledged. The first is the datasets used in the analysis are limited to 14 cancer types excluding pancreatic cancer. In the future, we will perform argonaute-2 photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation (AGO2-PAR-CLIP) in pancreatic cancer cells to generate a biochemically validated set of miRNA-binding sites. The second is the shortage of lncRNA expression microarray data in pancreatic cancer in public datasets. We only identified 21 differentially expressed lncRNAs, so that none of lncRNA was matched to expression data in lncRNA–miRNA–mRNA regulatory network. Due to this point, investigation of lncRNA expression profiles of pancreatic cancer and screen differentially expressed lncRNA are urgently needed. Finally, it should be emphasized that the two regulation mechanisms analyzed in this study were only bioinformatically predicted, thus should be considered for further validation and functional examination with in vivo and in vitro experiments.
References
Ascano, M., et al. (2012). Identification of RNA–protein interaction networks using PAR-CLIP. Wiley Interdisciplinary Reviews: Rna, 3(2), 159–177.
Cline, M. S., et al. (2007). Integration of biological networks and gene expression data using Cytoscape. Nature Protocols, 2(10), 2366–2382.
Darnell, R. B. (2010). HITS-CLIP: panoramic views of protein–RNA regulation in living cells. Wiley Interdisciplinary Reviews: Rna, 1(2), 266–286.
Dayem, U. A., et al. (2014). The pancreatic expression database: Recent extensions and updates. Nucleic Acids Research, 42, D944–D949.
Donahue, T. R., et al. (2012). Integrative survival-based molecular profiling of human pancreatic cancer. Clinical Cancer Research, 18(5), 1352–1363.
Farnham, P. J. (2009). Insights from genomic profiling of transcription factors. Nature Reviews Genetics, 10(9), 605–616.
Frampton, A. E., et al. (2014). MicroRNAs cooperatively inhibit a network of tumor suppressor genes to promote pancreatic tumor growth and progression. Gastroenterology, 146(1), 268–277.
Huang, D. W., Sherman, B. T., & Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols, 4(1), 44–57.
Jalali, S., et al. (2013). Systematic transcriptome wide analysis of lncRNA–miRNA interactions. PLoS One, 8(2), e53823.
Kefas, B., et al. (2010). Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro Oncology, 12(11), 1102–1112.
Kim, K., et al. (2013). HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene, 32(13), 1616–1625.
Konig, J., et al. (2011). Protein–RNA interactions: new genomic technologies and perspectives. Nature Reviews Genetics, 13(2), 77–83.
Li, J. H., et al. (2014). starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Research, 42(1), D92–D97.
Liu, J. H., et al. (2014). Expression and prognostic significance of lncRNA MALAT1 in pancreatic cancer tissues. Asian Pacific Journal of Cancer Prevention, 15(7), 2971–2977.
Ma, C., et al. (2014). H19 promotes pancreatic cancer metastasis by derepressing let-7′s suppression on its target HMGA2-mediated EMT. Tumor Biology, 1–7.
Panzitt, K., et al. (2007). Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology, 132(1), 330–342.
Park, P. J. (2009). ChIP-seq: advantages and challenges of a maturing technology. Nature Reviews Genetics, 10(10), 669–680.
Pepke, S., Wold, B., & Mortazavi, A. (2009). Computation for ChIP-seq and RNA-seq studies. Nature Methods, 6(11), S22–S32.
Tahira, A. C., et al. (2011). Long noncoding intronic RNAs are differentially expressed in primary and metastatic pancreatic cancer. Mol Cancer, 10, 141.
Wang, J., et al. (2010). CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Research, 38(16), 5366–5383.
Wang, G., et al. (2013). MicroRNA 125 represses nonsense-mediated mRNA decay by regulating SMG1 expression. Biochemical and Biophysical Research Communications, 435(1), 16–20.
Xu, H., et al. (2010). Liver-enriched transcription factors regulate microRNA-122 that targets CUTL1 during liver development. Hepatology, 52(4), 1431–1442.
Yang, J. H., et al. (2011). starBase: A database for exploring microRNA-mRNA interaction maps from argonaute CLIP-Seq and degradome-seq data. Nucleic Acids Research, 39, D202–D209.
Yang, J. H., et al. (2013). ChIPBase: A database for decoding the transcriptional regulation of long non-coding RNA and microRNA genes from ChIP-Seq data. Nucleic Acids Research, 41, D177–D187.
Zhao, Y., & Simon, R. (2008). BRB-ArrayTools Data Archive for human cancer gene expression: A unique and efficient data sharing resource. Cancer Informatics, 6, 9–15.
Zhao, W. G., et al. (2010). The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis, 31(10), 1726–1733.
Acknowledgments
This study was supported by The research Special Fund For public welfare industry of health (No. 201202007), and Fund for National key specialty construction of clinical project (General Surgery), and Fund from the Education Department of Zhejiang Province (No. Y201328225), and Fund from the Health Department of Zhejiang Province (No. 201484382), and National High Technology Research and Development Program of China (863 Program, No. 2012AA02A205), and the National Natural Science Foundation of China (No. J20121214), and the Financial Support of Science Technology Department of Zhejiang Province (No.2011C23088) and Medical Science Research Foundation of Health Bureau of Zhejiang Province (No. 2012KYB070).
Conflict of interest
We have no conflict of interest to declare and informed consent was obtained.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ye, S., Yang, L., Zhao, X. et al. Bioinformatics Method to Predict Two Regulation Mechanism: TF–miRNA–mRNA and lncRNA–miRNA–mRNA in Pancreatic Cancer. Cell Biochem Biophys 70, 1849–1858 (2014). https://doi.org/10.1007/s12013-014-0142-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-014-0142-y