
Abstract
Although many genes and miRNAs have been reported for various cancers, pancreatic cancer’s specific genes or miRNAs have not been studied precisely yet. Therefore, we have analyzed the gene and miRNA expression profile of pancreatic cancer data in the gene expression omnibus (GEO) database. The microarray-derived miRNAs and mRNAs were annotated by gene ontology (GO) and signaling pathway analysis. We also recognized mRNAs that were targeted by miRNA through the mirDIP database. An integrated analysis of the microarray revealed that only 6 out of 43 common miRNAs had significant differences in their expression profiles between the tumor and normal groups (P value < 0.05 and |log Fold Changes (logFC)|> 1). The hsa-miR-210 had upregulation, whereas hsa-miR-375, hsa-miR-216a, hsa-miR-217, hsa-miR-216b and hsa-miR-634 had downregulation in pancreatic cancer (PC). The analysis results also revealed 109 common mRNAs by microarray and mirDIP 4.1 databases. Pathway analysis showed that amoebiasis, axon guidance, PI3K-Akt signaling pathway, absorption and focal adhesion, adherens junction, platelet activation, protein digestion, human papillomavirus infection, extracellular matrix (ECM) receptor interaction, and riboflavin metabolism played important roles in pancreatic cancer. GO analysis revealed the significant enrichment in the three terms of biological process, cellular component, and molecular function, which were identified as the most important processes associated strongly with pancreatic cancer. In conclusion, DTL, CDH11, COL5A1, ITGA2, KIF14, SMC4, VCAN, hsa-mir-210, hsa-mir-217, hsa-mir-216a, hsa-mir-216b, hsa-mir-375 and hsa-mir-634 can be reported as the novel diagnostic or even therapeutic markers for the future studies. Also, the hsa-mir-107 and hsa-mir-125a-5p with COL5A1, CDH11 and TGFBR1 genes can be introduced as major miRNA and genes on the miRNA-drug-mRNA network. The new regulatory network created in our study could give a deeper knowledge of the pancreatic cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer (PC) is the 8th leading cause of death from cancer worldwide and the 4th most common cancer in the USA, with a mortality rate of about 30,000 deaths each year (Vincent et al. 2011; Pourshams et al. 2018; Han et al. 2015). Unfortunately, no definitive therapy has been found, and most of the patients die because of this disease (Pourshams et al. 2018). Modern tumor diagnosis and therapy is now looked at the molecular level ranging of the biomarkers (AbdelGhafar et al. 2019; El-Guindy et al. 2019; Tobin et al. 2015, Habib et al. 2020). To the best of our knowledge K-ras gene with over 85% of prevalence is one of the genes most involved in pancreatic cancers (Li et al. 2004; Uehara et al. 1999, Rungjarernarrejitt et al. 2018). P16, a tumor suppressor gene, as well as p53, are deactivated in about 95% of pancreatic cancers (Schutte et al. 1997). Imaging and marker analyses don't really discriminate between pancreatic adenocarcinoma and non-neoplastic pancreatic diseases in patients which pancreatic cancer is suspected. For these purposes, to promote early screening and identification, much work has been made to recognize reliable markers of pancreatic cancer (Goggins 2007). While some conventional markers such as cancer antigen (CA) 15-3 and carcinoembryonic antigen (CEA) are used for early screening and diagnosis of pancreatic cancer, its sensitivity and specificity are low (Ghafar et al. 2020). Other genes include HER2, MYB, AKT2, BRCA2, FHIT, CDKN2A, PALB2, STK11, and PRSS1, which are associated with the activation or inactivation of proto-oncogenes and other related genes, such as tumor suppressor genes. Although the new diagnosis and therapeutic approaches have grown over recent years (Rezaie et al. 2020a; Baudino 2015); the underlying causes of this cancer have not yet been fully identified (Han et al. 2015; Li et al. 2004; Hidalgo 2010). The microRNAs (miRNAs) are essential regulators of gene expression in cancer cells. The miRNAs are short, non-coding, and single-stranded RNA molecules (17–25-nucleotide), that can control gene expression, by joining to 3-untranslated region (3-UTR) genes or mRNA degradation (Andorfer et al. 2011; Schetter et al. 2008). The microRNAs participate in the regulation of cellular pathways such as apoptosis, differentiation, proliferation, metastasis, and senescence (Wang et al. 2015; Khafaei et al. 2019). Thus, they can be applied as potential therapeutic or diagnostic biomarkers in cancer (Andorfer et al. 2011; Schetter et al. 2008; Mendell and Olson 2012; Shah et al. 2016). They have been extremely involved in the pathogenesis of human cancer and some investigations showed that the miRNAs specifically act in various cancers. For example in breast cancer, miR-200 showed upregulation as an oncogene (Korpal et al. 2011), while in the lung, kidney, and ovarian cancers downregulation of miR-200 significantly correlated with decreased angiogenesis and poor survival (Pecot et al. 2013). In pancreatic cancer the hsa-miR-217, hsa-miR-96, miR-216a, and miR-148a/b by targeting KRAS and other genes, are downregulated, and also the miR-221, miR-210, miR-155, and miR-21 are upregulated (Xue et al. 2013). Nevertheless, in some cancers, microRNAs have dual functions. For example, the miR-221, which has been shown to be upregulated in liver and colon cancers, acts as an oncogenic factor by inhibiting tumor suppressors PTEN and RECK, whereas it acts as a tumor suppressor by derepression of oncogenes such as c-KIT and ETV1 (Berindan‐Neagoe et al. 2014). Although many miRNA and genes have been reported for various cancers (Berindan‐Neagoe et al. 2014; Sohrabi et al. 2020) such as miR-200c for ovarian cancer, hsa-miR-142-5p and hsa-miR-375 for gastric cancer, miR-133 for breast cancer, and miR-21 for colon cancer, and lung cancer, to best of our knowledge, for pancreatic cancer diagnosis, suitable markers with adequate accuracy are needed (Ghafar et al. 2020; Berindan‐Neagoe et al. 2014). Molecular markers are increasingly being suggested for the screening and diagnosis of PC, but they are not feasible for routine clinical use (Fry et al. 2008; Hasan et al. 2019).
Bioinformatics and system biology tools are remarkably used in the different fields of medical biology (Gasparini-Junior et al. 2019; Lu et al. 2018; Mohammadi et al. 2020; Rezaie et al. 2020b, 2018; Keshtvarz et al. 2017). Analyzing and understanding the genes-miRNAs network for cancer enables us to find genes and miRNAs that have significant expression level differences over cancer states. In addition, we can find genes significantly affected by miRNAs, identify different target genes for miRNAs, identify the mechanism of the disease, and finally introduce potential new and reliable biomarkers.
Aimed at gene expression analysis, the microarray provides a high-throughput platform that in recent years, has been extensively conducted as a powerful tool for assessing general genetic alteration during carcinogenesis (Kim 2004; Zhang et al. 2017). The microarray technology, which can calculate the expression changes of the different genes in the healthy and tumor groups, enables the researcher to study the expression of several thousands of genes simultaneously (Zhang et al. 2017). For example, in Yi lu study the ITGA2, MMP7, ITGB4, ITGA4, ITC3, VCAN, and PLAU genes have been identified after the bioinformatics analysis of the microarray data from patients with pancreatic ductal adenocarcinoma (Berindan‐Neagoe et al. 2014). Several studies also revealed that metabolic pathways and signaling interactions were associated with the shortening of life expectancy in this disease (Lu et al. 2018; Li et al. 2018). The MMP7 and ITGA2 genes have also been correlated with the prognosis and diagnosis of pancreatic cancer (Zhang et al. 2017; Li et al. 2018; Long et al. 2016). It is necessary to explore new molecular biomarkers facilitating detection or increasing the screening and treatment rate of pancreatic cancer. As a result, in the present work, we tried to study the genes-miRNAs-drugs network and understand their signaling pathways in pancreatic cancer to introduce the new diagnosis and therapeutic targets.
Methods
Pancreatic Cancer Biomarkers
We applied the medical subject headings (Rezaie et al. 2020a) based keywords (pancreatic cancer AND (biomarker* OR current marker*) AND discovery AND human) in Google Scholar, Pubmed from 2015 to 2020, and selected the most common biomarkers of pancreatic cancer.
Microarray Data
We searched expression profiles of miRNAs and mRNAs in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) using the following keywords: pancreatic neoplasms AND Homo sapiens. The inclusion criteria were as follows: (1) samples diagnosed with pancreatic cancer (PC) and normal tissue samples, (2) profiling of gene expression for mRNA and miRNA, and (3) enough data to plan the study.
Data Processing
GEO2R online webserver was used to identify differentially expressed genes and miRNAs between PC and normal tissue samples. As the criteria, a P value < 0.05 and a |logFC|> 1 were set. To increase the specificity, further analyses with common miRNAs in both datasets (P value < 0.05) were designed and submitted to the MirDIP database to obtain the miRNA’s target genes. Finally, we identified common genes between targets of miRNAs and differential expression genes (DEGs) using the Venn diagram.
Functional and Pathway Enrichment Analysis
Kyoto encyclopedia of genes and genomes (KEGG) and GO analysis, including biological process (BP), molecular function (Sandhu et al. 2016), and cellular component (Tahira et al. 2011), were used for functional study of single genes or large-scale genome. Both analyses were done in the EnrichR database (Chen et al. 2013; Kuleshov et al. 2016). The cut-off criterion was set to P value < 0.01.
Protein–Protein Interaction (PPI) Network Construction and Analysis of Modules
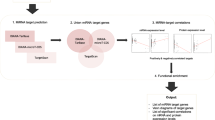
The DEGs were mapped to STRING (recovery tool for interacting genes) database (http://string-db.org/) to evaluate the protein–protein interaction integration and then visualized with Cytoscape. The molecular complex detection (MCODE) plug-in (http://apps.cytoscape.org/apps/mcode) was used to screen PPI network hub gene modules with cut-off degree = 10, haircut = on, node score cut-off = 0.2, k-core = 2, and max. Depth = 100. Clusters with more than 5 scores were selected for further analysis (Fig. 1).

Flow chart of data analysis. Firstly, we identified differentially expressed mRNAs and miRNAs in pancreatic cancer through integrating expression microarray data then we constructed network of miRNA–mRNA based on Microarray data. Also, we enrichment genes and analyzed protein–protein network and finally survival of genes were investigated
Survival Analysis
Survival analysis was performed using five studies with terms of Pancreatic Adenocarcinoma (TCGA, PanCancer Atlas), Pancreatic Adenocarcinoma (TCGA, Provisional), Pancreatic Cancer (UTSW, Nat Commun 2015), Pancreatic Adenocarcinoma (QCMG, Nature 2016) and Pancreatic Adenocarcinoma (ICGC, Nature 2012) from the cBioPortal database (Gao et al. 2013), (survival analysis refers to the overall survival Kaplan–Meier estimate). The datasets used in this study were composed of 1034 pancreatic cancer/adenocarcinoma samples/patients. Based on a relationship network built by MCODE (clusters 1 and 2 with score > 5), gene names were submitted in cBioPortal, and survival analysis was accomplished, of which genes with Log rank test of P < 0.05 were presented.
Construction of Regulatory miRNA-Drug-mRNA Network
Herein, we identified the mRNAs targeted by miRNA through mirDIP 4.1 (Tokar et al. 2017; Shirdel et al. 2011). Then, we identified the mRNAs that correlated with miRNA expression in pancreatic cancer through a Venn diagram. The target genes of the common miRNA from GSE71533 and GSE2479 were predicted with mirDIP, an online MicroRNA target-predicting database, that comprise of 28 database (BCmicrO,BiTargeting, CoMeTa, Cupid, DIANA,ElMMo3,GenMir++, microrna.org, mirbase, mirCoX, miRcode, miRDB, miRTar2GO, MAMI, MBStar, MirAncesTar, MirMAP, MirSNP, MirTar, Mirza-G, MultiMiTar, PACCMIT, PicTar, PITA, RepTar, RNA22, RNAhybrid, TargetRank, TargetScan and TargetSpy). The network was constructed between 43 selected miRNAs and 17 selected genes, including cluster one and two MCODE analysis (score > 5) in cytoscape version 3.6.0. Genes correlated with different miRNAs also entered into the DGIdb website (http://www.dgidb.org/) to find target gene drugs. A general network of genes, miRNAs, and their drugs was constructed with cytoscape software.
Results
Previous Biomarkers for Pancreatic Cancer
Some genes and miRNAs, which have been mentioned in previous studies as pancreatic cancer biomarkers, were identified. These genes and miRNAs are summarized in Table 1.
Many of these genes and miRNAs were excluded from our study due to insignificant P-Value and/or sharing between datasets (Table 2; Fig. 1). So far, many genes or miRNAs in pancreatic cancer had relatively good conditions for biomarkers, but often for various reasons, their importance has been diminished. Some of them have same expression profile in different cancers, others are not detectable in the early stages of cancer or at different stages. Others need to be studied with a larger sample size, some of which have only been tested in a pancreatic cancer subtype or under the influence of a particular drug, and some of them are not able to distinguish cancer from inflammatory conditions. These factors reduce their specificity and sensitivity.
Differential Expression Profiles of miRNAs and Genes
In this study after analysis of the 341 cases and 108 controls (Table 3), we tried to identify the miRNA and genes associated with pancreatic cancer and their interactions. Eventually, we could introduce the new potential biomarkers for this cancer. Efforts to understand the relationships between miRNAs and genes in different diseases already have been performed by the analysis of microarray data. After a search in the GEO database with the related keywords, five datasets (GSE15471, GSE16515, GSE30134, GSE46234, and GSE107610) were collected for gene expression and two datasets (GSE71533 and GSE24279) for miRNA expression analysis (Table 3).
Genes and miRNAs showing significant increase (P value < 0.05 and logFC > 1) and decrease (P value < 0.05 and logFC < − 1) were identified as DEGs and differential expression miRNAs (DEMs) (Fig. 1). Finally, as shown in Figs. 2 and 3, we reached the 309 DEGs (59 down and 250 upregulated distributed in 3 databases) and 6 DEMs (hsa-miR-210 upregulated and hsa-miR-217, hsa-miR-216a, hsa-miR-216b, hsa-miR-375, and hsa-miR-634 downregulated) (Fig. 3a, b). To increase the specificity and identify more genes, we performed further analysis with common miRNAs (43 numbers with P value < 0.05) in both datasets (Fig. 3c). They were submitted to the MirDIP database and 6006 genes were predicted for these miRNAs. The common genes between 309 (DEGs) and 6006 miRNA target genes were identified, which are shown in the Venn diagram. We ultimately obtained 109 mRNAs, of which 22 and 87 were down and upregulated respectively (Fig. 4). Unlike some previous studies and as it is shown in Fig. 1, there are some important criteria in our study such as presence in at least 3 datasets, over- or under-expression with P value < 0.05 and |logFC|> 1, the similarity with miRNAs targets, MCODE and survival analysis. These criteria have been caused to exclude many previously introduced biomarkers like CA19-9, TFF1, COL6A3 and CEACAM1 from our further analysis (Table 2).

Venn diagrams represent the commonly down and upregulated genes (309 DEGs), among the five identified databases. a Downregulated DEGs in pancreatic cancer among the five datasets were recognized via Venn diagrams, 59 genes were reported to be downregulated at least in three datasets. b Upregulated DEGs in pancreatic cancer among the five datasets, 250 genes were reported to be upregulated at least in three datasets

Venn diagrams represent common miRNAs with different filters identified in the two datasets. a DEMs with P value less than 0.05 and logFC < − 1 were identified in both datasets. b DEM with P value less than 0.05 and logFC > 1 were identified in both datasets. c Common miRNAs with P value less than 0.05 were identified in both datasets that were recognized via Venn diagrams

Venn diagrams shows common genes between 309 (DEGs) and 6006 (miRNA targets) genes were identified. Commonality between the miRNAs predicted targets (6006 genes) and common DEGs in at least 3 databases for down and upregulation genes (59 and 250 genes) which 22 and 87 genes were associated with down and upregulated genes respectively. Finally, enrichment for these genes (22 down and 87 up) was carried out with used EnrichR and their protein networks were constructed String database. Then, these protein networks were analyzed with MCODE in Cytoscape and the important genes were introduced
Functional and Pathway Enrichment Analyses
To further understand the functions and mechanisms of the identified DEGs, functional and pathway enrichment studies have been performed, including GO and KEGG, with the EnrichR database. The GO term enrichment analysis showed that the upregulated genes were massively enriched in the biological process-associated group like extracellular matrix organization, collagen fibril organization, skeletal system development, regulation of cell migration, regulation of cell proliferation, regulation of the apoptotic process, cellular response to cytokine stimulus, neutrophil degranulation and regulation of transcription, while the downregulated genes mainly involved in peptide metabolic process, cellular protein localization, cellular response to insulin stimulus and organonitrogen compound catabolic process (Table S1).
Also cell component analysis showed that the upregulated genes were enriched in the endoplasmic reticulum lumen, membrane rafts and tertiary granule, integral component of the plasma membrane, focal adhesion, and lysosome. The downregulation of the genes was mainly found in the integral component of the plasma membrane and endoplasmic reticulum-Golgi intermediate compartment (Table S2). Moreover, in terms of molecular function, the upregulated genes were enriched in platelet-derived growth factor binding, transcriptional activator activity, RNA polymerase II core promoter proximal region sequence-specific binding, collagen binding, kinase binding, peptidase activity, acting on l-amino acid peptides, and transcription regulatory region sequence-specific DNA binding. The downregulated genes were enriched in zinc ion binding, metalloaminopeptidase activity, transition metal ion binding, protein homodimerization activity, and aminopeptidase activity (Table S3).
Furthermore, the KEGG pathway analysis showed upregulated genes involved in ECM-receptor interaction, amoebiasis, Axon guidance, PI3K-Akt signaling pathway, Focal adhesion Protein digestion and absorption, adherens junction, Platelet activation, and human papillomavirus infection. Downregulated genes involved in riboflavin metabolism (Table 4).
Analysis of PPI Network Modules
Up- and downregulated DEGs in pancreatic cancer were mapped via the STRING database with a PPI score > 0.4. Twenty-two nodes and one edge for downregulated genes (Fig. 5a) and 87 nodes and 134 edges for upregulated genes were constructed (Fig. 5b). MCODE analysis cannot be used for downregulated genes due to the lack of network, but such analysis was constructed for three networks (Fig. 6a–c) with different scores, which are detailed in Table 5. Seventeen genes had scores > 5 after the MCODE analysis.

PPI networks of DEGs identified in pancreatic cancer status versus normal status. a Downregulation of DEGs identified in at least three datasets and common with DEMs target gene, were used to construct the PPI network. The lines between nodes represent the interactions between genes. b Upregulation of DEGs identified in at least three datasets and common with DEMs target gene. The color of each gene is determined based on the logFC. Guide for logFC are listed at the bottom right of the page

Three PPI modules were extracted from the PPI network using MCODE in Cytoscape. PPI protein–protein interaction, DEGs differentially expressed genes, blue nodes indicate upregulated genes. The color of each gene is determined based on the logFC. Guide for logFC are listed at the bottom right of the page
Survival Analysis
After MCODE analysis, seventeen genes have been assessed in cluster 1 and 2 for pancreatic cancer survival (derived from 5 studies by cBioPortal database). As shown in Table 6, among all genes, seven upregulated genes (CDH11, COL5A1, DTL, ITGA2, KIF14, SMC4, and VCAN) significantly shortened the life expectancy (P < 0.05) (Fig. 7).

Overall Survival Kaplan–Meier estimate of 7 genes in 1034 samples from TCGA datasets with term of Pancreatic Adenocarcinoma (TCGA, PanCancer Atlas), Pancreatic Adenocarcinoma (TCGA, Provisional), Pancreatic Cancer (UTSW, Nat Commun 2015), Pancreatic Adenocarcinoma (QCMG, Nature 2016) and Pancreatic Adenocarcinoma (ICGC, Nature 2012). Red line represents cases with alterations. Blue line represents cases without alterations (VCAN, P = 1.815e−3), (SMC4, P = 0.0146), (KIF14, P = 0.0161), (ITGA2, P = 6.090e−3), (DTL, P = 5.988e−5), (COL5A1, P = 0.0435), (CDH11, P = 0.0475)
This significant difference can be due to five datasets related to pancreatic cancer, the sharing of genes in at least three datasets, and having other applied criteria in this study (Fig. 1). We analyzed the survival of five previously identified genes (TFF1, COL6A3, NUSAP1, POSTN, and ARHGEF15), which were removed in our latest filter. Nevertheless, they did not seem to have significant effects on patients' survival length (log rank test P value > 0.05) (Fig. 8).

Overall survival Kaplan–Meier estimate of five genes that were previously identified as pancreatic cancer biomarkers and they removed in our latest filter in 1034 samples from TCGA datasets with term of Pancreatic Adenocarcinoma (TCGA, PanCancer Atlas), Pancreatic Adenocarcinoma (TCGA, Provisional), Pancreatic Cancer (UTSW, Nat Commun 2015), Pancreatic Adenocarcinoma (QCMG, Nature 2016) and Pancreatic Adenocarcinoma (ICGC, Nature 2012). Red line represents cases with alterations. Blue line represents cases without alterations (COL6A3, P = 0.725), (POSTN, P = 0.209), (ARHGEF15, P = 0.368). TFF1 and NUSAP1 did not have enough information in this database
miRNA-drug-mRNA Network Construction
Our goal in constructing this network is to identify important genes that are affected by different drugs and miRNAs that may be used as new agents in future studies of pancreatic cancer.
As shown in Fig. 9, initially, interaction analysis showed that multiple drugs were introduced for 12 genes (RACGAP1, ITGA2, COL5A1, COL3A1, COL1A2, COL1A1, CDH11, CCNA2, VCAN, TGFBR1, SDC1, and RRM2). Then the 23 miRNAs associated with these genes were identified, among those, 12 miRNAs (hsa-mir-107, hsa-mir-532-3p, hsa-mir-320a/b, hsa-mir-320c/d, hsa-mir-302e, hsa-mir-342-3p, hsa-mir-375, hsa-mir-217, hsa-mir-429 and hsa-mir-125a-5p) affects at least two gene.

Interaction network between the 17 gene from MCODE analysis (cluster one and two) and 43 reported miRNAs. Circle represent genes, rectangle represent miRNAs and round rectangle show drugs. Each miRNA and gene is recognized by a specific color and miRNAs with yellow rectangles have targets of more than 2 genes. 23 of 43 miRNA and 147 related drugs were associated with 17 genes, then these drugs were inserted into the network and the final form was constructed
Based on the figure, we found that Hsa-mir-107 and hsa-mir-125a-5p can regulate 6 genes in this network, that their common genes are COL5A1, CDH11, and TGFBR1. These two miRNA and three genes can be introduced as major miRNAs and genes of this network.
Discussion
The microarray data analysis has been used in many studies on the different types of tumors to find the important gene, miRNA, and metabolic pathways (Tu et al. 2019). In general, the role of miRNAs and genes in carcinogenesis and cancer progression is well known, but their expression, function, and association with pancreatic cancer development are not yet fully investigated (Sun et al. 2017). Early detection of small cancers before the onset of metastasis is currently the only tools to significantly improving resection, post-resection prognosis, and ultimately survival. However, not all available diagnostic tools and biomarkers for PDAC are able to detect early or preventive cancer and suffer from low specificity and sensitivity (Sharma et al. 2020).
In this study, we attempted to explore the interactions between genes, miRNAs, and important pathways in pancreatic cancer with the lowest filtration limitation and relatively high sample size through an accepted analysis method such as GEO2R. We identified the DTL, CDH11, COL5A1, ITGA2, KIF14, SMC4, and VCAN genes and hsa-mir-210, hsa-mir-217, hsa-mir-216a, hsa-mir-216b, hsa-mir-375, and hsa-mir-634 miRNAs associated with pancreatic cancer. Consequently, we could introduce the new potential biomarkers for this cancer.
One important gene is VCAN (belonging to the sulfate proteoglycans family), which has been expressed in many malignancies, such as pancreas, gastric, and prostate cancers. This gene often associated with poor prognosis (Barry et al. 2013; Jiang et al. 2019). In an integrated bioinformatics analysis for pancreatic ductal adenocarcinoma, some genes like MMP7, MMP1, COLA1, CDH11, COL3A1, ITGA2, and VCAN were introduced as hub genes. These may be potential targets for diagnosis, treatment, and prognostic prediction (Lu et al. 2018). Two studies conducted in 2014 and 2019 have shown that S100A6 was significantly upregulated in pancreatic carcinoma compared to normal tissues. They also showed that ITGA2 overexpression and hypomethylation of MET and ITGA2 were significantly associated with overall survival (Liu et al. 2019; Nones et al. 2014).
In compliance with our results, a study conducted by Mingge Shang et al. (2019 has shown an association between increased expression of six genes (KRT7, KRT19, SEMA3C, ITGA2, MYOF, and ANXA1) and their poor survival. PI3K-Akt, Focal adhesion and ECM-receptor interaction were also recognized as important pathways (Shang et al. 2019). It is noteworthy that these results were based on other databases and smaller samples than our investigation. Ivane Abiatari et al. (2009 showed upregulation of kinesin family member 14 (KIF14) and Rho-GDP dissociation inhibitor beta (ARHGDIbeta) mRNA levels in patients with pancreatic cancer (Abiatari et al. 2009).
Song Ye in 2014 analyzed the GEO-derived pancreatic cancer microarray data with 60 cancer samples and 21 controls. Through investigating the association between genes and miRNAs, they identified different miRNAs and genes as important elements in this cancer (Ye et al. 2014). In comparison to their study, the present research includes a higher number of samples and a new data analysis method. miRNA-210 had a significant upregulation in both studies, while miR-217 and miR-216a had a significant downregulation.
In agreement with our results, another study in 2014 on pancreatic cancer showed a significant increase in miR-210 and a significant decrease in miR-217, miR-216, and miR-375. Furthermore, our results showed a significant decrease in miR-216a and miR-634, but their expression has not been mentioned in that study (Ye et al. 2014). This difference can be due to the number of samples and the type of data analysis methods.
High expressions of miR-196a and miR-210 were associated with poor survival (P-Value = 0.001 and P value = 0.003) (Yu et al. 2017); however, miR-196a was excluded from our study due to lack of sharing between two miRNA datasets. miR-216a may potentially serve as a novel tumor marker and prognostic factor for pancreatic cancer and miR-217 functions as a prognosis predictor and inhibits pancreatic cancer cell proliferation, invasion and promotes apoptosis via targeting E2F3.
The next miRNA showing a significant downregulation in our study was miR-375. Previously, downregulation of this miRNA was demonstrated by in vitro methods such as real-time PCR in pancreatic, glioma, and gastric cancers. The hsa-miR-375 is associated with inhibition of migration and proliferation of pancreatic and glioma cancer cells via downregulating of RWDD3 gene (Ji et al. 2020). Our mRNA–miRNA interaction network also demonstrated that the hsa‑miR‑375 can regulate the RACGAP1, TGFBR1, SMC4, VCAN genes. The RACGAP1 gene is one of the most important predictors of poor prognosis in the pancreas adenocarcinoma (Khalid et al. 2019). TGFBR1 has been involved in invasion, migration, angiogenesis, and tumorigenesis of pancreatic cancer cells and it is also active in cellular pathways such as TGF-β/SMAD (Gasparini-Junior et al. 2019; Zhou et al. 2018).
Ultimately the last miRNA that was examined in the present study was miR-634, with a significant downregulation. This event has also been reported in different cancers (Gao et al. 2016; Fischer et al. 2001). Previous studies have already shown its anti-tumor role and promotive action in the apoptosis of cancer cells. The mRNA–miRNA interaction network showed that hsa‑miR-634 could regulate COL3A1. Upregulation of the COL3A1 gene is directly associated with gliomas, renal cell carcinoma (RCC), and bladder malignancies growth, metastasis, progression, and prognosis (Su et al. 2014; Shi and Tian 2019). It may be referred to as a diagnostic or therapeutic biomarker (Gao et al. 2016). In general, the inconsistency between the genes and miRNAs reported in the different system biology studies maybe depend on many factors such as the cancer type, the sample size, filtration approaches, and the procedures of data analysis.
These genes and miRNAs were found to be related to drug response. Inactivating mutations in both TGFBR1 and TGFBR2 have been observed in pancreatic carcinoma and constitutively decreased TGFBR1 expression may have a decreased risk of pancreatic cancer by regulating factors such as TGF-β-mediated growth inhibition (Adrian et al. 2009). In agreement with our results, in human pancreatic cancer (PC) samples and 12 PC cell lines, an increase in TGFBR1 expression has been observed (Adrian et al. 2009; Fan et al. 2020). Yue Fan and et al. (2020) showed that TGFBR1, VTCN1, and LGALS9 were found to be associated with the worse outcomes of patients with PC. Also, they demonstrated, upregulation of TGFBR1 was closely pertained to poor overall survival and significantly associated with the prognosis in PC (Fan et al. 2020).
Collagen type V (COL5) presents in most connective tissue matrix and plays a functional role in different cancers such as breast cancer (Barsky et al. 1982), colon cancer (Fischer et al. 2001), and pancreatic ductal adenocarcinoma (Berchtold et al. 2015). Bioinformatic identification showed that COL5A1 may be a major factor in many types of cancers like breast cancer, gastric cancer, papillary thyroid carcinoma, ovarian cancer, oral squamous cell carcinoma, and lung adenocarcinoma. Also, overexpression of COL5A1 significantly correlates with the overall survival of patients with clear cell renal cell carcinoma (Feng et al. 2019).
CDH11 signaling may play an essential role in mediating the function of activated stellate cells, defining the fibrotic and inflammatory microenvironment in both pancreatitis and cancer. In agreement with our results, Cadherin-11 (Cad-11, also known as OB cadherin or CDH11) has shown upregulation in pancreatic cancer and involved in pancreatic stellate cells (PSCs) activation and pancreatic cancer metastasis (Birtolo et al. 2017).
The miR-107 is abnormally expressed in several metastatic tumors like colorectal, breast, gastric, liver, lung, bladder, and cervical cancer, and its upregulation contributes to cancer progression and metastasis. Also, upregulation of miR-107 is associated with poor clinicopathological parameters and prognosis in pancreatic ductal adenocarcinoma (PDAC) patients (Xiong et al. 2017). miR‐107 had been considered as an oncogene miRNA in gastric and liver cancer (Gong et al. 2019).
miR-125a-5p has been shown to play critical functions in human malignancies. miR-125a-5p acted as a tumor suppressor in various human malignancies, such as hepatocellular carcinoma, breast cancer, lung cancer, gastric cancer, and glioblastoma (Gao et al. 2014). YANG and ZENG by integrating transcriptome analysis showed the upregulation of hsa-mir-125a-5p in miRNA–mRNA crosstalk in pancreatic cancer (Yang and Zeng 2015). Lichao Pan and et al. (2018) showed that the miR-125a by targeting mitofusin 2 (Mfn2), promotes apoptosis, metabolism disorder, and migration impairment in pancreatic cancer cells (Pan et al. 2018).
Conclusion
The important genes and miRNAs introduced in this study, due to the significant change in expression in tumor and normal tissue and the effect on patient survival, can be more specific and sensitive than previous tests for pancreatic cancer and may be used alone or in combination with other tests for diagnosis or prognosis. However, it is necessary for these identified genes and miRNAs to pass the experimental approvals later.
To the best of our knowledge, we attempted to identify DEGs using comprehensive bioinformatics evaluations and come up with potential biological markers to predict disease progression. After analysis, a total of 309 DEGs and 6 DEMs were screened with promising targets for pancreatic cancer treatment, management, and prognosis, among which the DTL, CDH11, COL5A1, ITGA2, KIF14, SMC4, VCAN, hsa-mir-210, hsa-mir-217, hsa-mir-216a, hsa-mir-216b, hsa-mir-375 and hsa-mir-634 had pivotal functions. Also, the wide relation of the hsa-mir-107 and hsa-mir-125a-5p and COL5A1, CDH11 and TGFBR1 genes to drug response was represented in the miRNA-drug-mRNA network. These outcomes call for more organized researches to model exact aspects of gene networks in pancreatic cancer.
References
Abdel Ghafar MT, Allam AA, Darwish S (2019) Serum HOX transcript antisense RNA expression as a diagnostic marker for chronic myeloid leukemia. Egypt J Haematol 44(2):91
Abiatari I, DeOliveira T, Kerkadze V, Schwager C, Esposito I, Giese NA et al (2009) Consensus transcriptome signature of perineural invasion in pancreatic carcinoma. Mol Cancer Ther 8(6):1494–1504
Adrian K, Strouch MJ, Zeng Q, Barron MR, Cheon EC, Honasoge A et al (2009) Tgfbr1 haploinsufficiency inhibits the development of murine mutant Kras-induced pancreatic precancer. Cancer Res 69(24):9169–9174
Andorfer CA, Necela BM, Thompson EA, Perez EA (2011) MicroRNA signatures: clinical biomarkers for the diagnosis and treatment of breast cancer. Trends Mol Med 17(6):313–319
Arafat H, Lazar M, Salem K, Chipitsyna G, Gong Q, Pan T-C et al (2011) Tumor-specific expression and alternative splicing of the COL6A3 gene in pancreatic cancer. Surgery 150(2):306–315
Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I (2008) Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia-the authors reported a combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology 55(88):2016
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M et al (2012) NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 41(D1):D991–D995
Barry S, Chelala C, Lines K, Sunamura M, Wang A, Marelli-Berg FM et al (2013) S100P is a metastasis-associated gene that facilitates transendothelial migration of pancreatic cancer cells. Clin Exp Metastas 30(3):251–264
Barsky SH, Rao C, Grotendorst GR, Liotta LA (1982) Increased content of type V collagen in desmoplasia of human breast carcinoma. Am J Pathol 108(3):276
Baudino TA (2015) Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol 12(1):3–20
Bauer AS, Keller A, Costello E, Greenhalf W, Bier M, Borries A et al (2012) Diagnosis of pancreatic ductal adenocarcinoma and chronic pancreatitis by measurement of microRNA abundance in blood and tissue. PLoS ONE 7(4):e34151
Berchtold S, Grünwald B, Krüger A, Reithmeier A, Hähl T, Cheng T et al (2015) Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett 356(2):721–732
Berindan-Neagoe I, Monroig PC, Pasculli B, Calin GA (2014) MicroRNAome genome: a treasure for cancer diagnosis and therapy. Cancer J Clin 64(5):311–336
Birtolo C, Pham H, Morvaridi S, Chheda C, Go VLW, Ptasznik A et al (2017) Cadherin-11 is a cell surface marker up-regulated in activated pancreatic stellate cells and is involved in pancreatic cancer cell migration. Am J Pathol 187(1):146–155
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV et al (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform 14(1):128
Chen L, Song H, Luo Z, Cui H, Zheng W, Liu Y et al (2020) PHLPP2 is a novel biomarker and epigenetic target for the treatment of vitamin C in pancreatic cancer. Int J Oncol 56(5):1294–1303
Deng Y, Ma H, Hao J, Xie Q, Zhao R (2020) MCM2 and NUSAP1 are potential biomarkers for the diagnosis and prognosis of pancreatic cancer. Bio Med Res Int. https://doi.org/10.1155/2020/8604340
Dong D, Jia L, Zhang L, Ma N, Zhang A, Zhou Y et al (2018) Periostin and CA242 as potential diagnostic serum biomarkers complementing CA19 9 in detecting pancreatic cancer. Cancer Sci 109(9):2841–2851
El-Guindy DM, Wasfy RE, Ghafar MTA, Ali DA, Elkady AM (2019) Oct4 expression in gastric carcinoma: association with tumor proliferation, angiogenesis and survival. J Egypt Natl Cancer Inst 31(1):3
Ellsworth KA, Eckloff BW, Li L, Moon I, Fridley BL, Jenkins GD et al (2013) Contribution of FKBP5 genetic variation to gemcitabine treatment and survival in pancreatic adenocarcinoma. PLoS ONE 8(8):e70216
Fan Y, Li T, Xu L, Kuang T (2020) Comprehensive analysis of immunoinhibitors identifies LGALS9 and TGFBR1 as potential prognostic biomarkers for pancreatic cancer. Comput Math Methods Med. https://doi.org/10.1155/2020/6138039
Feng G, Ma H-M, Huang H-B, Li Y-W, Zhang P, Huang J-J et al (2019) Overexpression of COL5A1 promotes tumor progression and metastasis and correlates with poor survival of patients with clear cell renal cell carcinoma. Cancer Manag Res 11:1263
Fischer H, Stenling R, Rubio C, Lindblom A (2001) Colorectal carcinogenesis is associated with stromal expression of COL11A1 and COL5A2. Carcinogenesis 22(6):875–878
Fry LC, Mönkemüller K, Malfertheiner P (2008) Molecular markers of pancreatic cancer: development and clinical relevance. Langenbecks Arch Surg 393(6):883–890
Fukushima H, Yasumoto M, Ogasawara S, Akiba J, Kitasato Y, Nakayama M et al (2016) ARHGEF15 overexpression worsens the prognosis in patients with pancreatic ductal adenocarcinoma through enhancing the motility and proliferative activity of the cancer cells. Mol Cancer 15(1):1–13
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6(269):p11
Gao W, Chan JY-W, Wong T-S (2014) Curcumin exerts inhibitory effects on undifferentiated nasopharyngeal carcinoma by inhibiting the expression of miR-125a-5p. Clin Sci 127(9):571–579
Gao Y-F, Mao X-Y, Zhu T, Mao C-X, Liu Z-X, Wang Z-B et al (2016) COL3A1 and SNAP91: novel glioblastoma markers with diagnostic and prognostic value. Oncotarget 7(43):70494
Gasparini-Junior JL, Fanelli MF, Abdallah EA, Chinen LTD (2019) Evaluating mmp-2 and tgfß-ri expression in circulating tumor cells of pancreatic cancer patients and their correlation with clinical evolution. Arq Bras Cir Dig. https://doi.org/10.1590/0102-672020190001e1433
Ghafar MTA, Gharib F, Al-Ashmawy GM, Mariah RA (2020) Serum high-temperature-required protein A2: a potential biomarker for the diagnosis of breast cancer. Gene Rep. https://doi.org/10.1016/j.genrep.2020.100706
Goggins M (2007) Identifying molecular markers for the early detection of pancreatic neoplasia. Semin Oncol 34(4):303–310
Golob-Schwarzl N, Puchas P, Gogg-Kamerer M, Weichert W, Göppert B, Haybaeck J (2020) New pancreatic cancer biomarkers eIF1, eIF2D, eIF3C and eIF6 play a major role in translational control in ductal adenocarcinoma. Anticancer Res 40(6):3109–3118
Gong J, Lu X, Xu J, Xiong W, Zhang H, Yu X (2019) Coexpression of UCA1 and ITGA2 in pancreatic cancer cells target the expression of miR-107 through focal adhesion pathway. J Cell Physiol 234(8):12884–12896
Goonesekere NC, Andersen W, Smith A, Wang X (2018) Identification of genes highly downregulated in pancreatic cancer through a meta-analysis of microarray datasets: implications for discovery of novel tumor-suppressor genes and therapeutic targets. J Cancer Res Clin Oncol 144(2):309–320
Habib EM, Nosiar NA, Eid MA, Taha AM, Sherief DE, Hassan AE et al (2020) Circulating miR-146a expression predicts early treatment response to imatinib in adult chronic myeloid leukemia. J Investig Med 69(2):333–337
Han DY, Fu D, Xi H, Li QY, Feng LJ, Zhang W et al (2015) Genomic expression profiling and bioinformatics analysis of pancreatic cancer. Mol Med Rep 12(3):4133–4140
Hasan S, Jacob R, Manne U, Paluri R (2019) Advances in pancreatic cancer biomarkers. Oncol Rev. https://doi.org/10.4081/oncol.2019.410
Hidalgo M (2010) Pancreatic cancer. N Engl J Med 362(17):1605–1617
Idichi T, Seki N, Kurahara H, Yonemori K, Osako Y, Arai T et al (2017) Regulation of actin-binding protein ANLN by antitumor miR-217 inhibits cancer cell aggressiveness in pancreatic ductal adenocarcinoma. Oncotarget 8(32):53180
Ji C-X, Fan Y-H, Xu F, Lv S-G, Ye M-H, Wu M-J et al (2018) MicroRNA-375 inhibits glioma cell proliferation and migration by downregulating RWDD3 in vitro. Oncol Rep 39(4):1825–1834. https://doi.org/10.3892/or.2020.7785
Jiang K, Liu H, Xie D, Xiao Q (2019) Differentially expressed genes ASPN, COL1A1, FN1, VCAN and MUC5AC are potential prognostic biomarkers for gastric cancer. Oncol Lett 17(3):3191–3202
Keshtvarz M, Salimian J, Yaseri M, Bathaie SZ, Rezaie E, Aliramezani A et al (2017) Bioinformatic prediction and experimental validation of a PE38-based recombinant immunotoxin targeting the Fn14 receptor in cancer cells. Immunotherapy 9(5):387–400
Khafaei M, Rezaie E, Mohammadi A, Shahnazi Gerdehsang P, Ghavidel S, Kadkhoda S et al (2019) miR-9: from function to therapeutic potential in cancer. J Cell Physiol 234(9):14651–14665
Khalid M, Idichi T, Seki N, Wada M, Yamada Y, Fukuhisa H et al (2019) Gene regulation by antitumor miR-204-5p in pancreatic ductal adenocarcinoma: the clinical significance of direct RACGAP1 regulation. Cancers 11(3):327
Kim H (2004) Role of microarray in cancer diagnosis. Cancer Res Treat 36(1):1
Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celià-Terrassa T et al (2011) Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med 17(9):1101
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44(W1):W90–W97
Li D, Xie K, Wolff R, Abbruzzese JL (2004) Pancreatic cancer. Lancet 363(9414):1049–1057
Li C, Zeng X, Yu H, Gu Y, Zhang W (2018) Identification of hub genes with diagnostic values in pancreatic cancer by bioinformatics analyses and supervised learning methods. World J Surg Oncol 16(1):223
Liu L, Wang S, Cen C, Peng S, Chen Y, Li X et al (2019) Identification of differentially expressed genes in pancreatic ductal adenocarcinoma and normal pancreatic tissues based on microarray datasets. Mol Med Rep 20(2):1901–1914
Liu H, Li X, Dun MD, Faulkner S, Jiang CC, Hondermarck H (2020) Cold shock domain containing E1 (CSDE1) protein is overexpressed and can be targeted to inhibit invasiveness in pancreatic cancer cells. Proteomics 20(10):1900331
Long J, Liu Z, Wu X, Xu Y, Ge C (2016) Gene expression profile analysis of pancreatic cancer based on microarray data. Mol Med Rep 13(5):3913–3919
Lu Y, Li C, Chen H, Zhong W (2018) Identification of hub genes and analysis of prognostic values in pancreatic ductal adenocarcinoma by integrated bioinformatics methods. Mol Biol Rep 45(6):1799–1807
Luo G, Liu C, Guo M, Cheng H, Lu Y, Jin K et al (2017) Potential biomarkers in Lewis negative patients with pancreatic cancer. Ann Surg 265(4):800–805
Makawita S, Dimitromanolakis A, Soosaipillai A, Soleas I, Chan A, Gallinger S et al (2013) Validation of four candidate pancreatic cancer serological biomarkers that improve the performance of CA19. 9. BMC Cancer 13(1):404
Mantini G, Vallés A, Le Large T, Capula M, Funel N, Pham T et al (2020) Co-expression analysis of pancreatic cancer proteome reveals biology and prognostic biomarkers. Cell Oncol 43(6):1147–1159
Mao Y, Shen J, Lu Y, Lin K, Wang H, Li Y et al (2017) RNA sequencing analyses reveal novel differentially expressed genes and pathways in pancreatic cancer. Oncotarget 8(26):42537
Mendell JT, Olson EN (2012) MicroRNAs in stress signaling and human disease. Cell 148(6):1172–1187
Mohammadi M, Rezaie E, Sakhteman A, Zarei N (2020) A highly potential cleavable linker for tumor targeting antibody-chemokines. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1841025
Nones K, Waddell N, Song S, Patch AM, Miller D, Johns A et al (2014) Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int J Cancer 135(5):1110–1118
Pan L, Zhou L, Yin W, Bai J, Liu R (2018) miR-125a induces apoptosis, metabolism disorder and migrationimpairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission. Int J Oncol 53(1):124–136
Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C et al (2013) Tumour angiogenesis regulation by the miR-200 family. Nat Commun 4:2427
Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W et al (2009) FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16(3):259–266
Pourshams A, Kazemi B, Kalantari S (2018) A review of the etiology and biomarkers of pancreatic cancer, with emphasis on the role of diabetes. Tehran Univ Med J 75(11):773–778
Radon TP, Massat NJ, Jones R, Alrawashdeh W, Dumartin L, Ennis D et al (2015) Identification of a three-biomarker panel in urine for early detection of pancreatic adenocarcinoma. Clin Cancer Res 21(15):3512–3521
Rajamani D, Bhasin MK (2016) Identification of key regulators of pancreatic cancer progression through multidimensional systems-level analysis. Genome Med 8(1):38
Rezaie E, Mohammadi M, Sakhteman A, Bemani P, Ahrari S (2018) Application of molecular dynamics simulations to design a dual-purpose oligopeptide linker sequence for fusion proteins. J Mol Model 24(11):313
Rezaie E, Amani J, Pour AB, Hosseini HM (2020a) A new scfv-based recombinant immunotoxin against EPHA2-overexpressing breast cancer cells; High in vitro anti-cancer potency. Eur J Pharmacol 870:172912
Rezaie E, Bidmeshki Pour A, Amani J, Mahmoodzadeh HH (2020b) Bioinformatics predictions, expression, purification and structural analysis of the PE38KDEL-scfv immunotoxin against EPHA2 receptor. Int J Pept Res Ther 26(2):979–996
Rückert F, Pilarsky C, Grützmann R (2010) Serum tumor markers in pancreatic cancer—recent discoveries. Cancers 2(2):1107–1124
Rungjarernarrejitt S, Hlaing E, Piamrojanaphat P, Lailerd N. Effect of purple rice bran in proteomic profile of pancreas of diabetic pancreas. Innovation of Functional Foods in Asia (IFFA). 2018, p 153
Sahni S, Krisp C, Molloy MP, Nahm C, Maloney S, Gillson J et al (2020) PSMD11, PTPRM and PTPRB as novel biomarkers of pancreatic cancer progression. Biochem Biophys Acta. https://doi.org/10.1016/j.bbagen.2020.129682
Sahni S, Krisp C, Molloy MP, Nahm C, Maloney S, Gillson J et al (2020) PSMD11, PTPRM and PTPRB as novel biomarkers of pancreatic cancer progression. Biochim Biophys Acta. https://doi.org/10.1016/j.bbagen.2020.129682
Sandhu V, Lothe IB, Labori K, Skrede M, Hamfjord J, Dalsgaard A et al (2016) Differential expression of miRNAs in pancreatobiliary type of periampullary adenocarcinoma and its associated stroma. Mol Oncol 10(2):303–316
Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N et al (2008) MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA 299(4):425–436
Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK et al (1997) Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Can Res 57(15):3126–3130
Seino T, Kawasaki S, Shimokawa M, Tamagawa H, Toshimitsu K, Fujii M et al (2018) Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 22(3):454–467
Shah MY, Ferrajoli A, Sood AK, Lopez-Berestein G, Calin GA (2016) microRNA therapeutics in cancer—an emerging concept. EBioMedicine 12:34–42
Shams R, Saberi S, Zali M, Sadeghi A, Ghafouri-Fard S, Aghdaei HA (2020) Identification of potential microRNA panels for pancreatic cancer diagnosis using microarray datasets and bioinformatics methods. Sci Rep 10(1):1–15
Shang M, Zhang L, Chen X, Zheng S (2019) Identification of hub genes and regulators associated with pancreatic ductal adenocarcinoma based on integrated gene expression profile analysis. Discov Med 28(153):159–172
Sharma GG, Okada Y, Von Hoff D, Goel A (2020) Non-coding RNA biomarkers in pancreatic ductal adenocarcinoma. Semin Cancer Biol. https://doi.org/10.1016/j.semcancer.2020.10.001
Shi S, Tian B (2019) Identification of biomarkers associated with progression and prognosis in bladder cancer via co-expression analysis. Cancer Biomark 24(2):183–193
Shirdel EA, Xie W, Mak TW, Jurisica I (2011) NAViGaTing the micronome–Using multiple microRNA prediction databases to identify signalling pathway-associated microRNAs. PLoS ONE 6(2):e17429
Simeone DM, Ji B, Banerjee M, Arumugam T, Li D, Anderson MA et al (2007) CEACAM1, a novel serum biomarker for pancreatic cancer. Pancreas 34(4):436–443
Sogawa K, Takano S, Iida F, Satoh M, Tsuchida S, Kawashima Y et al (2016) Identification of a novel serum biomarker for pancreatic cancer, C4b-binding protein α-chain (C4BPA) by quantitative proteomic analysis using tandem mass tags. Br J Cancer 115(8):949–956
Sohrabi E, Moslemi M, Rezaie E, Nafissi N, Afkhami H, Khaledi M et al (2020) The tissue expression of MCT3, MCT8, and MCT9 genes in women with breast cancer. Eur PMC. https://doi.org/10.21203/rs.3.rs-104529/v1
Su B, Zhao W, Shi B, Zhang Z, Yu X, Xie F et al (2014) Let-7d suppresses growth, metastasis, and tumor macrophage infiltration in renal cell carcinoma by targeting COL3A1 and CCL7. Mol Cancer 13(1):206
Sun H, Zhao L, Pan K, Zhang Z, Zhou M, Cao G (2017) Integrated analysis of mRNA and miRNA expression profiles in pancreatic ductal adenocarcinoma. Oncol Rep 37(5):2779–2786
Svoronos C, Tsoulfas G, Souvatzi M, Chatzitheoklitos E (2020) Prognostic value of COL6A3 in pancreatic adenocarcinoma. Ann Hepatobiliary Pancreat Surg 24(1):52–56
Tahira AC, Kubrusly MS, Faria MF, Dazzani B, Fonseca RS, Maracaja-Coutinho V et al (2011) Long noncoding intronic RNAs are differentially expressed in primary and metastatic pancreatic cancer. Mol Cancer 10(1):141
Tobin NP, Foukakis T, De Petris L, Bergh J (2015) The importance of molecular markers for diagnosis and selection of targeted treatments in patients with cancer. J Intern Med 278(6):545–570
Tokar T, Pastrello C, Rossos AE, Abovsky M, Hauschild A-C, Tsay M et al (2017) mirDIP 4.1—integrative database of human microRNA target predictions. Nucleic Acids Res 46(D1):D360–D370
Tu J, Chen J, He M, Tong H, Liu H, Zhou B et al (2019) Bioinformatics analysis of molecular genetic targets and key pathways for hepatocellular carcinoma. Onco Targets Ther 12:5153
Uehara H, Nakaizumi A, Tatsuta M, Baba M, Takenaka A, Uedo N et al (1999) Diagnosis of pancreatic cancer by detecting telomerase activity in pancreatic juice: comparison with K-ras mutations. Am J Gastroenterol 94(9):2513
Vincent A, Herman J, Schulick R, Hruban RH, Goggins M (2011) Pancreatic cancer. Lancet 378(9791):607–620
Wang B, Shen Z-l, Jiang K-W, Zhao G, Wang C-y, Yan Y-c et al (2015) MicroRNA-217 functions as a prognosis predictor and inhibits colorectal cancer cell proliferation and invasion via an AEG-1 dependent mechanism. BMC Cancer 15(1):437
Wei J, Yang L, Wu Y-n, Xu J (2020) Serum miR-1290 and miR-1246 as potential diagnostic biomarkers of human pancreatic cancer. J Cancer 11(6):1325
Wei Y, Wang G, Wang C, Zhou Y, Zhang J, Xu K (2020) Upregulation of DUSP14 affects proliferation, invasion and metastasis, potentially via epithelial-mesenchymal transition and is associated with poor prognosis in pancreatic cancer. Cancer Manag Res 12:2097
Wu J, Wang Y, Jiang Z (2020) TNFSF9 is a prognostic biomarker and correlated with immune infiltrates in pancreatic cancer. J Gastrointest Cancer. https://doi.org/10.1007/s12029-020-00371-6
Xiao D, Dong Z, Zhen L, Xia G, Huang X, Wang T et al (2020) Combined exosomal GPC1, CD82, and serum CA19-9 as multiplex targets: a specific, sensitive, and reproducible detection panel for the diagnosis of pancreatic cancer. Mol Cancer Res 18(2):300–310
Xie Z, Yin X, Gong B, Nie W, Wu B, Zhang X et al (2015) Salivary microRNAs show potential as a noninvasive biomarker for detecting resectable pancreatic cancer. Cancer Prev Res 8(2):165–173
Xiong J, Wang D, Wei A, Lu H, Tan C, Li A et al (2017) Deregulated expression of miR-107 inhibits metastasis of PDAC through inhibition PI3K/Akt signaling via caveolin-1 and PTEN. Exp Cell Res 361(2):316–323
Xue Y, Tayoun ANA, Abo KM, Pipas JM, Gordon SR, Gardner TB et al (2013) MicroRNAs as diagnostic markers for pancreatic ductal adenocarcinoma and its precursor, pancreatic intraepithelial neoplasm. Cancer Genet 206(6):217–221
Yang J, Zeng Y (2015) Identification of miRNA-mRNA crosstalk in pancreatic cancer by integrating transcriptome analysis. Eur Rev Med Pharmacol Sci 19(5):825–834
Ye S, Yang L, Zhao X, Song W, Wang W, Zheng S (2014) Bioinformatics method to predict two regulation mechanism: TF–miRNA–mRNA and lncRNA–miRNA–mRNA in pancreatic cancer. Cell Biochem Biophys 70(3):1849–1858
Ye Z-Q, Zou C-l, Chen H-b, Jiang M-j, Mei Z, Gu D-n (2020) MicroRNA-7 as a potential biomarker for prognosis in pancreatic cancer. Disease Mark. https://doi.org/10.1155/2020/2782101
Yu Q, Xu C, Yuan W, Wang C, Zhao P, Chen L et al (2017) Evaluation of plasma microRNAs as diagnostic and prognostic biomarkers in pancreatic adenocarcinoma: miR-196a and miR-210 could be negative and positive prognostic markers, respectively. Biomed Res Int. https://doi.org/10.1155/2017/6495867
Zhang Y, Han X, Wu H, Zhou Y (2017) Bioinformatics analysis of transcription profiling of solid pseudopapillary neoplasm of the pancreas. Mol Med Rep 16(2):1635–1642
Zhou B, Guo W, Sun C, Zhang B, Zheng F (2018) Linc00462 promotes pancreatic cancer invasiveness through the miR-665/TGFBR1-TGFBR2/SMAD2/3 pathway. Cell Death Dis 9(6):1–15
Zhou Q, Andersson R, Hu D, Bauden M, Kristl T, Sasor A et al (2019) Quantitative proteomics identifies brain acid soluble protein 1 (BASP1) as a prognostic biomarker candidate in pancreatic cancer tissue. EBioMedicine 43:282–294
Zhou Q, Bauden M, Andersson R, Hu D, Marko-Varga G, Xu J et al (2020) YAP1 is an independent prognostic marker in pancreatic cancer and associated with extracellular matrix remodeling. J Transl Med 18(1):1–10
Zhu H, Wei M, Xu J, Hua J, Liang C, Meng Q et al (2020) PARP inhibitors in pancreatic cancer: molecular mechanisms and clinical applications. Mol Cancer 19(1):1–15
Acknowledgements
The authors would like to thank office center of Baqiyatallah Research Center for Gastroenterology and Liver Diseases for providing the right conditions to doing this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors are not declaring any conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sohrabi, E., Rezaie, E., Heiat, M. et al. An Integrated Data Analysis of mRNA, miRNA and Signaling Pathways in Pancreatic Cancer. Biochem Genet 59, 1326–1358 (2021). https://doi.org/10.1007/s10528-021-10062-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-021-10062-x