
Abstract
Fatty acid–binding protein 3 (FABP3) facilitates the movement of fatty acids in cardiac muscle. Previously, we reported that FABP3 is highly upregulated in the myocardium of ventricular septal defect patients and overexpression of FABP3 inhibited proliferation and promoted apoptosis in embryonic carcinoma cells (P19 cells). In this study, we aimed to investigate the effect of FABP3 gene silencing on P19 cell differentiation, proliferation and apoptosis. We used RNA interference and a lentiviral-based vector system to create a stable FABP3-silenced P19 cell line; knockdown of FABP3 was confirmed by quantitative real-time PCR. Expression analysis of specific differentiation marker genes using quantitative real-time PCR and observation of morphological changes using an inverted microscope revealed that knockdown of FABP3 did not significantly affect the differentiation of P19 cells into cardiomyocytes. CCK-8 proliferation assays and cell cycle analysis demonstrated that FABP3 gene silencing significantly inhibited P19 cell proliferation. Furthermore, Annexin V-FITC/propidium iodide staining and the caspase-3 activity assay revealed that FABP3 gene silencing significantly promoted serum starvation–induced apoptosis in P19 cells. In agreement with our previous research, these results demonstrate that FABP3 may play an important role during embryonic heart development, and that either overexpression or silencing of FABP3 will lead to an imbalance between proliferation and apoptosis, which may result in embryonic cardiac malformations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart development is controlled by an evolutionarily conserved network of transcription factors which connect the signaling pathways and genes responsible for muscle growth, patterning and contractility, and involves a precisely orchestrated series of molecular and morphogenetic events. Mutations in components of the cardiac gene network or subtle perturbation of the processes which these genes regulate can have catastrophic consequences in the form of congenital heart disease (CHD) [1]. CHD is currently the most common type of birth defect, with an incidence ranging from approximately 4 to 50 per 1,000 live births [2, 3]. Although the mortality rate of CHD has decreased due to advances in medical and surgical management, not all CHD patients survive to adulthood.
Ventricular septal defect (VSD) is the most common form of CHD and among the most frequently observed congenital abnormalities [4]. The ontogeny of ventricular septation is complex and requires coordinated proliferation, specification, differentiation, migration, apoptosis, morphogenesis and cellular interactions [5]. As a multifactorial complex disease, a number of environmental and genetic factors have been identified to play an important role in the occurrence of VSD. With the recent development of new techniques in molecular and developmental biology, the past decade has witnessed spectacular progress in elucidation of the molecular mechanisms involved in CHD. However, so far, very few of the genes which cause VSD have been identified. Therefore, the identification and characterization of novel genes and proteins associated with VSD remains an issue of intense interest.
Fatty acid–binding protein 3 (FABP3) is a low molecular weight protein with a distinct tissue-specific distribution pattern, which plays an important role in fatty acid transport, cell growth, cellular signaling and gene transcription [6, 7]. In previous studies using suppression subtractive hybridization [8], we found that FABP3 is highly expressed in the myocardium of VSD patients. Further studies revealed that overexpression of FABP3 inhibited proliferation and promoted apoptosis in embryonic myocardial cells and affected the differentiation of cardiac precursors into mature cardiomyocytes [9]. Therefore, in the present study, we aimed to investigate whether FABP3 gene silencing is capable of enhancing proliferation, inhibiting apoptosis and affecting the differentiation of embryonic myocardial cells. The P19 cell line is a suitable model for cardiac differentiation at the molecular and functional levels [10]; therefore, we selected P19 cells to investigate the impact of FABP3 gene silencing on embryonic myocardial cell differentiation, proliferation and apoptosis.
Materials and Methods
Cell line and Cell Culture
P19 cells (embryonic carcinoma cells) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells (the sixth generation) were cultured in α-modified Eagle’s medium (α-MEM; Gibco BRL, Grand Island, NY, USA) supplemented with 10 % fetal bovine serum (FBS; Gibco BRL), 100 U/ml penicillin and 100 μg/ml streptomycin at 37 °C in 5 % CO2. In order to induce cardiac differentiation, P19 cells were cultivated as aggregates from days 0 to 4 in the presence of α-MEM containing 10 % FBS and 1 % DMSO in bacteriological dishes at 37 °C in 5 % CO2, as previously described [11]. At day 4, the embryoid bodies were transferred to 6-cm culture flasks and then cultured in α-MEM containing 10 % FBS for an additional 4 or 6 days. The morphological changes in P19 cells were examined and photographed using an inverted microscope (Nikon, Japan).
Construction of RNAi Lentiviral Vector
A short hairpin RNA (shRNA) construct was designed to contain a unique 19-nt double-stranded FABP3 sequence presented as an inverted complementary repeat, a loop sequence (5′-CTCGAG-3′), the RNA PloIII terminator (5′-TTTTTT-3′) and 5′ single-stranded overhangs for ligation into AgeI- and EcoRI-digested pGLV–U6–Puro lentivirus vector (GenePharma, Shanghai, China). The recombinant vector was named pGLV–FABP3–shRNA. The negative control vector (pGLV–NC–shRNA) contained a nonsense shRNA insert in order to control for effects due to non-RNAi mechanisms. The sequences of the two cDNA fragments (positive-sense strand) were as follows: FABP3, 5′-CATCCTGACTCTCACTCAT-3′; negative control, 5′-TTCTCCGAACGTGTCACGT-3′.
Virus Generation and Infection
The lentiviral supernatant was produced by transient co-transfection of HEK-293 T cells with the pGLV–FABP3–shRNA or pGLV–NC–shRNA lentiviral plasmids with three optimized packaging plasmids: a packing plasmid (pGag/Pol), a VSV-G-expressing plasmid (pVSV-G) and a Rev (pRev)-expressing plasmid, using the method of Chen and Okayama [12]. The supernatants were collected 48 h after transfection, filtered through a 0.45 μm membrane, supplemented with hexadimethrine bromide (polybrene) to a final concentration of 5 μg/ml and applied directly to P19 cells. Stably transduced cells were selected using puromycin, by adding the minimum concentration of puromycin required to kill untransduced cells. The efficiency of knockdown was analyzed by quantitative real-time polymerase chain reaction (qPCR) using the primers shown in Table 1.
Quantitative Real-Time Reverse Transcription PCR
Quantitative real-time reverse transcription PCR (qRT-PCR) was performed to determine the expression of marker genes during P19 cell differentiation. Total RNA was extracted from P19 cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and cDNA was synthesized from 1 μg total RNA using the AMV Reverse Transcriptase Kit (Promega, Madison, WI, USA). Real-time PCR was performed using the SYBR green method in an Applied Biosystems 7300 Sequence Detection System (ABI 7300 SDS; Foster City, CA, USA), following the manufacturer’s protocols. The sequences of the primers used are shown in Table 1. The PCR conditions included a denaturation step (95 °C for 10 min), followed by 40 cycles of amplification and quantification (95 °C for 15 s, 60 °C for 1 min). Relative gene expression levels were quantified based on the cycle threshold (C t ) values and normalized to the reference gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Each sample was measured in triplicate, and the gene expression levels were calculated by the 2−ΔΔ C t method, as described previously [13].
Cell Growth Assay
Cell growth was analyzed using the WST-8 Cell Counting Kit-8 (Dojindo, Tokyo, Japan) according to the manufacturer’s instructions. Briefly, the cells were seeded in 96-well plates and incubated in α-MEM supplemented with 10 % FBS for 7 days. On the day of growth measurement, 100 μl of the spent media was replaced with an equal volume of fresh media containing 10 % CCK-8, and the cultures were incubated at 37 °C for 120 min. Absorbance was measured at 450 nm using a microplate reader. The results were plotted as the mean ± SD of three separate experiments with three determinations per experiment for each experimental condition.
Cell Cycle Analysis
Cell cycle distribution was analyzed using flow cytometry. Before analysis, the cells were serum deprived for 24 h to synchronize the cell cycle, then the cells were incubated with α-MEM containing 10 % FBS for 24 h after serum deprivation. The cells were trypsinized, rinsed with phosphate-buffered saline (PBS), fixed with 70 % ethanol at 4 °C overnight and treated with RNaseA (0.02 mg/ml) in the dark at room temperature for 30 min. The cells were resuspended in 0.05 mg/ml propidium iodide (PI; Sigma, St. Louis, MO, USA) and analyzed by flow cytometry (BD Biosciences, USA). At least 1 × 104 cells were recorded for each sample, and the data were analyzed using Cell Quest software (BD Biosciences).
Quantification of Apoptosis
Cells were cultured in serum-free α-MEM for 24 h to induce apoptosis, and then cultured in complete medium containing 10 % FBS for 0 or 24 h, harvested using trypsin, washed with PBS, centrifuged, resuspended in 1 ml binding buffer and stained with 10 μl Annexin V–FITC and 10 μl PI at room for 15 min (Biovision, California, CA, USA). The stained cells were analyzed immediately using flow cytometry as previously described [14]. Cell Quest software was used for data acquisition and analysis, and the percentage of apoptotic cells was determined.
Measurement of Caspase-3 Activity
Cells were cultured in serum-free α-MEM for 24 h to induce apoptosis, then collected and washed with PBS. Caspase-3 activity was assayed using a caspase-3 colorimetric assay kit (KeyGen, Nanjing, China) according to the manufacturer’s protocol. Briefly, 5 × 106 cells were lysed in 50 μl chilled cell lysis buffer on ice for 20 min, centrifuged for 1 min at 10,000 rpm at 4 °C. Aliquots of the supernatant containing 200 μg protein were diluted to 50 μl with cell lysis buffer, incubated with 5 μl substrate at 37 °C for 4 h, and the absorbance value of the samples were read at 405 nm in a microtiter plate reader.
Statistical Analysis
Each experiment was performed at least three times. The data are presented as the mean ± standard deviation (SD) values. All data were analyzed by the Student’s t test using the SPSS 13.0 statistical package (SPSS, Chicago, Il, USA); P < 0.05 was considered statistically significant.
Results
Creation of A Stable FABP3-silenced P19 Cell Line
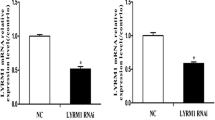
The knockdown of FABP3 was evaluated by real-time PCR. The relative expression of FABP3 mRNA in P19 cells was normalized to the endogenous control gene GAPDH. As shown in Fig. 1, P19 cells transfected with lentiviral FABP3-shRNA expressed significantly lower levels of FABP3 mRNA than the cells transfected with the negative control (NC) lentiviral construct (*P < 0.05). The steady state FABP3 mRNA expression level in the lentiviral-FABP3-shRNA-transfected cells was 76 % of NC cells.

Quantitative RT-PCR confirmation of FABP3 gene silencing in pGLV–FABP3–shRNA-transduced P19 cells. FABP3 mRNA expression was inhibited by 76 % in P19 cells transfected with pGLV–FABP3–shRNA (FABP3 RNAi), compared to cells transduced with control pGLV–NC–shRNA lentivirus (NC). Values are the mean ± SD of three independent experiments (*P < 0.05)
FABP3 Gene Silencing has No Effect on the Morphology of P19 Cells During Differentiation
To investigate the effect of FABP3 gene silencing on the differentiation of P19 cells into cardiac cells, we examined the morphological changes in P19 cells during DMSO-induced differentiation. The morphology of P19 cell on days 0, 4 and 10 of differentiation are shown in Fig. 2. The cardiac myocardial cells aggregated on day 4 of differentiation, and beating cell colonies appeared on day 10 of differentiation. However, no differences were observed in the morphological changes or time taken for the appearance of beating cell clusters in NC cells and lentiviral-FABP3-shRNA-transfected cells.

FABP3 gene silencing has no effect on the morphology of P19 cells during P19 cell cadio-differentiation. P19 cells were transfected with pGLV–FABP3–shRNA (FABP3 RNAi) or pGLV–NC–shRNA lentivirus (NC) and treated with 1 % DMSO to induce cardiomyocyte differentiation over 10 days. No obvious morphological differences were observed in FABP3-silenced and NC cells at days 0, 4 and 10
FABP3 Gene Silencing has No Effect on the Expression of Marker Genes During P19 Cell Cadio-differentiation
ES (embryonic stem) cell-derived cardiomyocytes express cardiac-specific genes and proteins in a developmentally controlled manner. For example, induction of cardiac troponin T (cTnT) mimics the isoform transition which occurs during cardiogenesis in vivo. The early cardiac transcription factor Nkx2.5, GATA4 and the cardiac muscle-specific marker α-MHC [15] were used as cardiac-specific markers in this study. We used qRT-PCR to analyze the expression levels of these genes during differentiation. As shown in Fig. 3, the expression of all the marker genes gradually increased during the process of differentiation; however, no significant difference in the mRNA expression levels of any marker gene was observed in FABP3-silenced cells and NC cells at any time point (days 0, 6, 8 and 10; P > 0.05).

FABP3 gene silencing has no effect on the expression of marker genes during P19 cardiomyocyte differentiation. P19 cells were transfected with pGLV–FABP3–shRNA (FABP3 RNAi) or pGLV–NC–shRNA lentivirus (NC) and treated with 1 % DMSO to induce cardiomyocyte differentiation, as described in the Materials and Methods. The expression of specific differentiation markers (cTnT, GATA4, Nkx2.5 and α-MHC) was determined by quantitative real-time PCR at various time points during differentiation (days 0, 6, 8 and 10). Values are the mean ± SD of three independent experiments (n = 6; P > 0.05)
FABP3 Gene Silencing Reduces P19 Cell Proliferation
The growth of FABP3-silenced and NC P19 cells was assessed using the CCK-8 cell viability assay. As shown in Fig. 4a, FABP3-silenced cells had a reduced growth rate compared to NC control cells. At days 1, 2 and 3, the OD values of FABP3-silenced and NC control cells were not significantly different. However, from day 4 onwards, the OD values of FABP3-silenced cells were significantly lower than control cells (*P < 0.05).

FABP3 gene silencing reduces P19 cell proliferation. P19 cells were transfected with pGLV–FABP3–shRNA (FABP3 RNAi) or pGLV–NC–shRNA lentivirus (NC). Cell growth was quantified using the CCK-8 cell viability assay (a) and cell cycle analysis (b). Values are the mean ± SD of three independent experiments (*P < 0.05)
We also performed flow cytometry analysis of the cell cycle distribution in FABP3-silenced and NC control P19 cells, and observed that the proportion of S phase cells in FABP3-silenced P19 cells was much lower than NC control cells (Fig. 4b).
FABP3 Gene Silencing Induces P19 Cell Apoptosis
Annexin V–FITC binds to phosphatidylserine which can be used to detect the early stages of apoptosis [14]. Apoptosis was induced by serum starvation for 24 h; then, the cells were cultured in complete media for 0 or 24 h, and the numbers of apoptotic cells were quantified by flow cytometric analysis after staining with Annexin V–FITC/PI. FABP3 gene silencing increased the number of apoptotic cells in response to serum deprivation (*P < 0.05; Fig. 5a, b). Additionally, the caspase-3 activity assay (Fig. 5c) also indicated that FABP3 gene silencing increased the number of apoptotic cells in response to serum deprivation. These results indicate that FABP3 gene silencing promotes serum deprivation-induced apoptosis.

FABP3 gene silencing induces P19 cell apoptosis in response to serum starvation. P19 cells were transfected with pGLV–FABP3–shRNA (FABP3 RNAi) or pGLV–NC–shRNA lentivirus (NC), serum starved for 24 h, then cultured in complete media for 0 or 24 h. Apoptosis was quantified by flow cytometry using Annexin V-FITC/PI staining (a, b) and the caspase-3 activity assay (c). Values are the mean ± standard deviation of three independent experiments (n = 6, *P < 0.05)
Discussion
Observational epidemiological studies have demonstrated the existence of genetic predispositions for CHD. Although many genes have been found to be important for cardiac development in animal studies, to date, only a few human genes have been associated with CHD as critical regulators of heart formation, including NKX2.5 [16], Tbx5 [17] and GATA4 [18]. Moreover, for most types of CHD, the basic pathogenetic mechanisms of VSD remain poorly characterized.
Previous studies have shown that FABP3 is expressed and upregulated in mouse neonatal hearts during cardiomyocyte terminal differentiation [19]. In present study, we used the P19 cell line as a research model, by establishing a stable line of FABP3-silenced P19 cells. RNA interference (RNAi) is a form of posttranscriptional gene silencing mediated by short double-stranded RNAs, known as small interfering RNAs (siRNA). RNAi is a powerful gene silencing mechanism which operates in most eukaryotic cells [20]. Long-lasting RNAi-based gene silencing can be achieved with the help of lentivirus-based expression systems, which drive the production of short hairpin RNA species (shRNAs) under the control of the RPPH1 (encoding ribonuclease PRNA component H1) promoter [21–23]. Moreover, lentiviral vectors encoding antisense targeting sequences have been used in clinical trials with no obvious side effects [22]. In this study, we successfully constructed an RNAi lentiviral vector to suppress endogenous expression of FABP3. The siRNA expression vector carrying antibiotics mark we employed can continuously inhibit the expression of target genes for several weeks in cells. Using real-time PCR technology, we confirmed that FABP3 gene silencing resulted in a significant reduction in FABP3 transcription. These effects were not observed in cells infected with the negative control shRNA.
We observed that knockdown of FABP3 did not affect the differentiation of P19 cells into cardiomyocytes, as indicated by the morphological changes and expression of cardiomyogenesis-specific molecular markers. However, FABP3 gene silencing dramatically inhibited proliferation and promoted apoptosis in P19 cells. In an earlier study, we also found that overexpression of FABP3 inhibited proliferation, promoted apoptosis and affected the differentiation of cardiac precursors into mature cardiomyocytes [24], which suggests that overexpression or silencing of FABP3 may influence morphogenesis in the embryonic heart by inducing excessive apoptosis in the myocardium.
In normal physical state, FABP3 is expressed quantitatively in cardiac muscle tissue, involving in energy metabolism as translocators of long-chain fatty acid to intracellular organelles. So, whether ectopic expression or FABP3-RNAi will lead to the destruction of the physiological balance, and further influence myocardial energy metabolism which can induce mitochondrial impairment that further promote apoptosis [25, 26].
The heart is the first organ to form in the embryo, and a complex series of morphogenetic interactions involving cells from several embryonic origins is involved in the development of this organ [27]. Embryonic fetal heart growth depends on the balance between myocyte proliferation and apoptosis [28]. Inadequate proliferation or excess apoptosis can directly or indirectly result in CHD [29], which is often caused by altered proliferation and/or apoptosis in the septum, neighboring tissue or myocardium [30]. Our results demonstrate that knockdown of FABP3 inhibited proliferation and promoted apoptosis in embryonic myocardial cells. We speculate that the molecular mechanisms underlying this effect may be related to mitochondrial impairment as a result of FABP3 gene silencing. FABP3 participates in mitochondrial energy metabolism by transporting the fatty acids produced during the process of myocardial cell ATP generation. In addition, the mitochondrion has been regarded as a key element in apoptosis, as it plays a core role in the regulation of mammalian apoptosis [31]. We hypothesize that FABP3 gene silencing downregulated FABP3 protein expression, which affected energy metabolism in the cells and subsequently lead to mitochondrial dysfunction which, in turn, triggered apoptosis.
P19 cells, derived from an early mouse embryonic tumor, are pluripotent in nature and can differentiate into various cell types depending on the environment [32]. Cellular aggregation is known to be a requirement for the DMSO-induced differentiation of P19 cells into cardiac myocytes. Efficient differentiation of P19 cells depends on the prior formation of non-adherent aggregates which resemble the inner cell mass of an embryo; however, the molecular events that occur during aggregation and which are necessary for cardiac differentiation are not fully understood [10]. In this study, both FABP3-silenced and control P19 cells could be induced to differentiate into cardiac cells, as indicated by observation of the typical morphological changes during differentiation. Simultaneously, we also investigated the expression of cardiac muscle-specific molecular markers (cTnT, GATA4, Nkx2.5 and α-MHC) during differentiation using quantitative real-time PCR. However, there was no significant difference in the mRNA expression levels of these marker genes in FABP3-silenced cells and NC cells at days 0, 6, 8 and 10. Thus, we suggest that FABP3 may play a role in the regulation of cardiac cell proliferation and apoptosis; however, FABP3 is not required for the differentiation of cardiac precursors into mature cardiomyocytes.
In summary, our results demonstrate that FABP3 silencing promotes apoptosis and inhibits the proliferation of P19 cells. On the basis of previous research [9], our results suggest that both overexpression and silencing of FABP3 can lead to an imbalance between proliferation and apoptosis, which may result in embryonic cardiac malformations. This study indicates that FABP3 may be a potential target gene which could be modulated to prevent VSD. However, the detailed molecular mechanisms and signaling pathways which mediate the imbalance between proliferation and apoptosis in response to FABP3 gene silencing or overexpression require further investigation in future studies.
References
Olson, E. N. (2006). Gene regulatory networks in the evolution and development of the heart. Science, 313, 1922–1927.
Hoffman, J. I., & Kaplan, S. (2002). The incidence of congenital heart disease. Journal of the American College of Cardiology, 39, 1890–1900.
Garg, V. (2006). Insights into the genetic basis of congenital heart disease. Cellular and Molecular Life Sciences, 63, 1141–1148.
Sands, A. J., Casey, F. A., Craig, B. G., Dornan, J. C., Rogers, J., & Mulholland, H. C. (1999). Incidence and risk factors for ventricular septal defect in “low risk” neonates. Archives of Disease in Childhood. Fetal and Neonatal Edition, 81, F61–F63.
Kaynak, B., von Heydebreck, A., Mebus, S., Seelow, D., Hennig, S., Vogel, J., et al. (2003). Genome-wide array analysis of normal and malformed human hearts. Circulation, 107, 2467–2474.
Besnard, P., Niot, I., Poirier, H., Clement, L., & Bernard, A. (2002). New insights into the fatty acid-binding protein (FABP) family in the small intestine. Molecular and Cellular Biochemistry, 239, 139–147.
Qian, Q., Kuo, L., Yu, Y. T., & Rottman, J. N. (1999). A concise promoter region of the heart fatty acid-binding protein gene dictates tissue-appropriate expression. Circulation Research, 84, 276–289.
Zhang, H., Zhou, L., Yang, R., Sheng, Y., Sun, W., Kong, X., et al. (2006). Identification of differentially expressed genes in human heart with ventricular septal defect using suppression subtractive hybridization. Biochemical and Biophysical Research Communications, 342, 135–144.
Zhu, C., Hu, D. L., Liu, Y. Q., Zhang, Q. J., Chen, F. K., Kong, X. Q., et al. (2011). Fabp3 inhibits proliferation and promotes apoptosis of embryonic myocardial cells. Cell Biochemistry and Biophysics, 60, 259–266.
van der Heyden, M. A., van Kempen, M. J., Tsuji, Y., Rook, M. B., Jongsma, H. J., & Opthof, T. (2003). P19 embryonal carcinoma cells: A suitable model system for cardiac electrophysiological differentiation at the molecular and functional level. Cardiovascular Research, 58, 410–422.
Grepin, C., Nemer, G., & Nemer, M. (1997). Enhanced cardiogenesis in embryonic stem cells overexpressing the GATA-4 transcription factor. Development, 124, 2387–2395.
Chen, C., & Okayama, H. (1987). High-efficiency transformation of mammalian cells by plasmid DNA. Molecular and Cellular Biology, 7, 2745–2752.
Livak, K. J., & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods, 25, 402–408.
Vermes, I., Haanen, C., Steffens-Nakken, H., & Reutelingsperger, C. (1995). A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. Journal of Immunological Methods, 184, 39–51.
Koike, M., Sakaki, S., Amano, Y., & Kurosawa, H. (2007). Characterization of embryoid bodies of mouse embryonic stem cells formed under various culture conditions and estimation of differentiation status of such bodies. Journal of Bioscience and Bioengineering, 104, 294–299.
Lints, T. J., Parsons, L. M., Hartley, L., Lyons, I., & Harvey, R. P. (1993). Nkx-2.5: A novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development, 119, 419–431.
Bruneau, B. G., Nemer, G., Schmitt, J. P., Charron, F., Robitaille, L., Caron, S., et al. (2001). A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell, 106, 709–721.
Watt, A. J., Battle, M. A., Li, J., & Duncan, S. A. (2004). GATA4 is essential for formation of the proepicardium and regulates cardiogenesis. Proceedings of the National Academy of Sciences of the United States of America, 101, 12573–12578.
Tang, M. K., Kindler, P. M., Cai, D. Q., Chow, P. H., Li, M., & Lee, K. K. (2004). Heart-type fatty acid binding proteins are upregulated during terminal differentiation of mouse cardiomyocytes, as revealed by proteomic analysis. Cell and Tissue Research, 316, 339–347.
Aagaard, L., & Rossi, J. J. (2007). RNAi therapeutics: Principles, prospects and challenges. Advanced Drug Delivery Reviews, 59, 75–86.
Dykxhoorn, D. M., Novina, C. D., & Sharp, P. A. (2003). Killing the messenger: Short RNAs that silence gene expression. Nature Reviews Molecular Cell Biology, 4, 457–467.
Abbas-Terki, T., Blanco-Bose, W., Deglon, N., Pralong, W., & Aebischer, P. (2002). Lentiviral-mediated RNA interference. Human Gene Therapy, 13, 2197–2201.
Brummelkamp, T. R., Bernards, R., & Agami, R. (2002). A system for stable expression of short interfering RNAs in mammalian cells. Science, 296, 550–553.
Zhu, C., Hu, D. L., Liu, Y. Q., Zhang, Q. J., Chen, F. K., Kong, X. Q., et al. (2011). Fabp3 inhibits proliferation and promotes apoptosis of embryonic myocardial cells. Cell Biochemistry and Biophysics, 60, 259–266.
Shen, Y. H., Song, G. X., Liu, Y. Q., Sun, W., Zhou, L. J., Liu, H. L., et al. (2012). Silencing of FABP3 promotes apoptosis and induces mitochondrion impairment in embryonic carcinoma cells. Journal of Bioenergetics and Biomembranes, 44, 317–323.
Song, G. X., Shen, Y. H., Liu, Y. Q., Sun, W., Miao, L. P., Zhou, L. J., Liu, H. L., Yang, R., Kong, X. Q., Cao, K. J., Qian, L. M., & Sheng, Y. H. (2012). Overexpression of FABP3 promotes apoptosis through inducing mitochondrial impairment in embryonic cancer cells. Journal of Cellular Biochemistry, Jul 2. doi: 10.1002/jcb.24243. [Epub ahead of print].
Srivastava, D. (2006). Genetic regulation of cardiogenesis and congenital heart disease. Annual Review of Pathology: Mechanisms of Disease, 1, 199–213.
Fiorina, P., Corradi, D., Pinelli, S., Maestri, R., Lagrasta, C., Buscaglia, M., et al. (2004). Apoptotic/mytogenic pathways during human heart development. International Journal of Cardiology, 96, 409–417.
Levy, M., Maurey, C., Celermajer, D. S., Vouhe, P. R., Danel, C., Bonnet, D., et al. (2007). Impaired apoptosis of pulmonary endothelial cells is associated with intimal proliferation and irreversibility of pulmonary hypertension in congenital heart disease. Journal of the American College of Cardiology, 49, 803–810.
Gittenberger-de, G. A., Bartelings, M. M., Deruiter, M. C., & Poelmann, R. E. (2005). Basics of cardiac development for the understanding of congenital heart malformations. Pediatric Research, 57, 169–176.
Hoye, A. T., Davoren, J. E., Wipf, P., Fink, M. P., & Kagan, V. E. (2008). Targeting mitochondria. Accounts of Chemical Research, 41, 87–97.
McBurney, M. W., Jones-Villeneuve, E. M., Edwards, M. K., & Anderson, P. J. (1982). Control of muscle and neuronal differentiation in a cultured embryonal carcinoma cell line. Nature, 299, 165–167.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81070138), the Natural Science Foundation of Jiangsu Province, China (No. BK2010582) and the Talent Foundation of Jiangsu Province, China (WSN-020).
Author information
Authors and Affiliations
Corresponding author
Additional information
Yahui Shen, Guixian Song contributed equally to this work.
Rights and permissions
About this article
Cite this article
Shen, Y., Song, G., Liu, Y. et al. Silencing of FABP3 Inhibits Proliferation and Promotes Apoptosis in Embryonic Carcinoma Cells. Cell Biochem Biophys 66, 139–146 (2013). https://doi.org/10.1007/s12013-012-9462-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-012-9462-y