
Abstract
We determined the effect of atorvastatin on myocardial apoptosis and caspase-8 activation following coronary microembolization (CME) in a rat model. For this, 50 rats were randomly and equally divided into CME; sham-operated (control); atorvastatin lavage; gastric lavage control; and caspase-8 inhibitor (CHO) groups. In CME animals, a microembolization ball was injected through the left ventricle. Sham animals were injected with normal saline (NS). Atorvastatin group received atorvastatin gastric lavage once-a-day, 1 week before surgery. Gastric lavage controls had similar lavage with NS. CHO group was i.p-injected (CHO: 10 mg/kg) 30 min before surgery. Cardiac indices in each group were determined by echocardiography 6-h postoperatively. TUNEL assay and western blot were used for myocardial apoptosis and expression of caspases-3/-8, respectively. Echocardiography data show that left ventricular ejection fraction (LVEF) in CME group was significantly decreased (P < 0.05) compared with sham controls. Besides, left ventricular fractional shortening (FS) and cardiac output (CO) were also decreased with an increase in left ventricular end-diastolic dimension (LVEDd). Atorvastatin and CHO animals had significantly improved (P < 0.05) cardiac function compared with CME group. Myocardial apoptosis and activation levels of caspases-3/-8 were significantly increased (P < 0.05) compared with sham; myocardial apoptosis and activation levels of caspases-3/-8 were significantly decreased (P < 0.05) in atorvastatin and CHO groups compared with CME group. In conclusion, atorvastatin pretreatment suppressed post-CME myocardial apoptosis and improved cardiac function through the blockade of a myocardial death receptor-mediated apoptotic pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coronary microembolization (CME) is a common complication following rupture of an atherosclerotic plaque during acute coronary syndrome (ACS) and functions as a bridge to vascular intervention. The incidence of CME is approximately 15–20%, increasing up to 30–45% during procedures such as great saphenous vein grafts or percutaneous coronary intervention (PCI). CME causes transient “no blood flow” or “slow flow” [1, 2] and is an independent predictor of poor long-term prognosis in patients who have suffered from acute myocardial infarction [3]. Once CME occurs, intracoronary thrombolytics, nitroglycerin, or platelet glycoprotein GPIIb/IIIa receptor antagonists along with remedial measures of direct mechanical embolus removal cannot improve the short- or long-term prognosis of patients. Consequently, CME remains a clinical dilemma on a routine basis [4].
Clinical trials have shown that atorvastatin (Lipitor) treatment 12 h before PCI can significantly improve the outcome in ACS patients and decrease by 88% the risk of 30-day major adverse cardiac events (MACEs) such as myocardial infarction, target-vessel revascularization, and death [5]. It was reported that daily use of atorvastatin (40 mg) for 7 days before PCI significantly reduced the perioperative myocardial injury in patients with stable angina [6]. It has been also shown that in patients suffering from myocardial ischemia reperfusion injuries, statins play a role against inflammation and apoptosis by reducing cytokine release, expression of cell adhesion molecules, and neutrophil aggregation [7]. It is known that myocardial apoptosis occurs following CME [8] and statins can inhibit the development of myocardial apoptosis. However, the potential role of statins in the treatment of CME is not fully understood. Therefore, we sought to determine the effect of atorvastatin pretreatment on myocardial apoptosis and activation of caspase-8 (a key apoptotic protein involved in death receptor pathways) using a rodent model of CME. In this article, we report that atorvastatin pretreatment suppresses post-CME myocardial apoptosis and improves cardiac function through the blockade of a myocardial death receptor-mediated apoptosis pathway.
Materials and Methods
Materials
Microembolization balls (lyophilized powder) were obtained from the Dynal Corporation, Norway. These balls had a diameter of 42 μM, with ~2 × 107 balls in each gram of powder, providing a final density of 3 × 104 balls/ml. The Terminal-deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) Kit was purchased from American Roche, and atorvastatin (Lipitor) was obtained from Pfizer. Rabbit anti-rat caspase-3 polyclonal antibody (pAb), mouse anti-rat caspase-8 monoclonal antibody (mAb), capsase-8 inhibitor Ac-IETD-CHO and horse radish peroxidase (HPR)-labeled mouse anti-GADPH mAb were purchased from Cell Signaling Technology, Inc. Danvers, MA (USA), Santa Cruz Biotechnology Inc, Santa Cruz, CA (USA), Calbiochem/EMD Chemicals, Gibbstown, NJ (USA) and Shanghai KangCheng (China), respectively.
Experimental Animals
A total of 50 healthy adult male Sprague–Dawley (SD) rats, weighing 200–250 g were purchased from the Center for Experimental Animals, Guangxi Medical University, Nanning City, China. Rats were housed under constant conditions at a temperature of 23 ± 1°C, humidity of 40 ± 5% and maintained on a 12-h light/dark cycle with ad libitum access to pellet food and tap water. Animal experiments were performed according to the European Community Guidelines for the Care and Use of Animals, and the research protocol was approved by the institutional ethics committee for animals’ use.
Modeling and Grouping
Rats were randomly assigned to the CME, sham control, atorvastatin lavage, gastric lavage control, and CHO groups, 10 rats in each group. The CME model used was as previously described [9]. In brief, the rats were anesthetized with 10% chloral hydrate (3 ml/kg body weight) i.p., and the trachea was intubated to maintain spontaneous respiration with a respirator. The chest was opened, and the ascending aorta was isolated. Then, 3,000 microspheres (Dynal, Oslo, Norway) suspended in 0.1 ml of physiological saline were quickly injected into the left ventricle during a 10-s occlusion of the ascending aorta. The occlusion device was removed after injection, and the chest was sutured after the heart rate and breathing returned to normal. Using the same procedure, sham control animals were instead administered an injection of 0.1 ml of physiological saline. The atorvastatin group received a daily gastric lavage with atorvastatin powder dissolved in physiological saline (20 mg/Kg/day) for 1 week before embolism. An injection of an equivalent amount of physiological saline was used in the gastric lavage control group. The CHO group was administered CHO (10 mg/kg body weight) i.p. For this purpose, CHO was dissolved in phosphate buffered saline (PBS) containing 2% dimethyl sulfoxide (DMSO) [10] 30 min before surgery. All other groups were injected with an equivalent amount of DMSO.
Cardiac Function
Six hours following CME, the rats were anesthetized, and the left ventricle was visualized using echocardiography. In brief, a 10 MHz transducer was placed on the left anterior chest wall to record the measures of LVEF, LVEDd, FS, and CO. End-diastolic volume (EDV) and end systolic volume (ESV) of the left ventricle were calculated using Simpson’s method [11]. LVEF and FS were calculated as follows:
Echocardiography was performed using a Philips Sonos 7500 system (Philips, Andover, USA). Evaluation of cardiac function at each time point was performed in triplicate, with the average reported.
Tissue Sampling
Following assessment of cardiac function, 10% KCl (2–3 ml) was injected into the caudal vein causing cessation of heart beat during diastole. Later, cardiectomy was performed, the atrial appendage and atrium cordis were removed, and the ventricle was segmented into the apical and base portions from the left ventricle long axis midpoint along the direction parallel to the atrioventricular groove. The apical portion of the heart was frozen in liquid nitrogen and stored at −80°C for western blot analysis. The base portion was fixed in 4% paraformaldehyde for 12 h, embedded in paraffin, and sectioned into at least 12 slices, each 4 μm thick. Three pieces containing the same number of microinfarction foci were selected for assessment of apoptosis using TUNEL assay. Hematoxylin and eosin (H&E) and hematoxylin-basic fuchsin-picric acid (HBFP) stainings were also preformed in triplicate to detect the area of myocardial microinfarction.
Myocardial Microinfarction Area
To detect the area of the infarct, paraffin sections were stained using HBFP method as previously described which stains red for the infarcted myocardium and yellow or brown for the normal myocardium. Five microscopic visual fields (×100) were randomly examined from each slice sample, and DMR + Q550 pathological image pattern analysis was used to examine HBFP-stained slices. Leica Qwin analysis software (Germany) and the planar area method were used to measure the infarct zone. Data are expressed as the average percent area of bulk analysis in the sample slice [8].
Myocardial Apoptosis
Myocardial apoptosis was determined using TUNEL assay and following the manufacturer’s instructions. Microscopic apoptotic nuclei (TUNEL positive) were stained yellow brown. A total of 40 non-overlapping zones (400×) from each sample slice were randomly observed within the microinfarction zone, the marginal zone, and the zone distant from the infarct [12]. Myocardial apoptotic number and overall myocardial cell number were counted, and the myocardial apoptotic rate was calculated as follows: Apoptotic number/Overall cell number × 100.
Western Blot
To detect the levels of activated caspase-3 and -8 in the myocardial tissue, antibodies directed against shear-activated caspase-3 and -8 were used in western immunoblots. Total myocardial protein was extracted using lysis buffer, and Lowry method was used to determine protein concentration. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed using 10% gel and 50 μg of protein per well. Polyvinylidene fluoride (PVDF) membrane transfer was conducted at 100 mA current for 2 h. The membrane was then incubated in blocking solution at room temperature (RT) for 1 h, rinsed with tris-buffered saline and tween-20 (TBST), and incubated with rabbit anti-rat caspase-3 pAb (1:1,000) or mouse anti-rat caspase-8 mAb (1:400) at RT for 2 h. HRP-labeled anti-GADPH (1:1,000) was used as the internal loading control. After rinsing, the membrane was incubated with the corresponding secondary antibodies for 1 h. Bio-Rad Gel Doc 2000 imaging system and software were used to calculate the integrated absorbance (IA) of identified bands as follows: IA = Area × Average density. After normalizing to GAPDH levels, the ratios of caspase-3 and -8 IA to GADPH IA were used to represent the relative levels of activated caspase-3 and -8.
Statistical Analysis
Data were expressed as mean ± standard deviation (SD). Comparisons were made using Student’s t-test or the analysis of variance (ANOVA) followed by Tukey HSD test. For linear correlation analysis, the Pearson’s correlation coefficient (r) was computed using SPSS software (SPSS, Chicago, Illinois, USA). Statistical significance was set at the P-value of <0.05.
Results
Animal Groups
No statistically significant differences (P > 0.05) were found in body weight or heart rate between all groups (data not shown).
Changes in Cardiac Function
Cardiac ultrasound performed 6 h postoperatively showed that the CME and lavage control groups had significantly decreased cardiac function as compared to that of sham group (Table 1). This outcome was characterized by systolic dysfunction and left ventricular dilation with significant decreases in LVEF, FS, and CO; however, LVEDd was found to be significantly increased (P < 0.05). Pretreatment with atorvastatin improved all indices of cardiac function in CME group. Specifically, the LVEF, FS, and CO were significantly increased, and LVEDd was significantly decreased (P < 0.05) as compared to lavage control group. CHO was able to improve the microembolism-associated cardiac dysfunction. CHO group had significantly higher LVEF, FS, and CO and lower LVEDd as compared with CME group (P < 0.05).
CME Histopathology
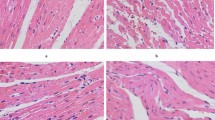
As revealed by H&E and HBFP stainings, the sham control animals had subendocardial ischemia without infarction foci. The CME, lavage control, CHO, and atorvastatin groups all had multiple microinfarction foci. Most of the foci were wedge-shaped, locally distributed, and non-transmural, and were observed mostly in the subendocardium and left ventricle (Fig. 1). H&E staining revealed the myocardial karyolysis or hypochromatosis with the cytoplasmic red dyeing in the microinfarction foci. In addition, peripheral cardiac muscle edema and denaturation, peripheral inflammatory cell infiltration, and erythrocyte effusion were also observed (Fig. 2). Infarct areas in the CME, lavage control, CHO, and atorvastatin groups were 8.32 ± 3.27, 8.21 ± 3.01, 7.88 ± 2.72, and 7.62 ± 2.58, respectively, with no significant difference found among the groups.

Histopathology of myocardial microinfarcts. HBFP staining of tissue samples revealed no infarct in the sham-operated (sham control; #1) animals. Microinfarcts in the tissue samples from CME (#2), lavage control (#3), CHO (#4), and atorvastatin (#5) groups are indicated by arrows. Normal myocardial cytoplasm is stained yellow, the nuclei are stained blue, and the ischemic myocardium is stained red (HBFP staining, ×100) (Color figure online)

Histopathology of post-CME myocardial microinfarcts. H&E staining of tissue samples from the coronary microembolization group reveals the myocardial microinfarcts with a low level of inflammatory cell infiltration. However, a portion of the cardiomyocyte nuclei is not visible. The arrow indicates the presence of a 42 μm microsphere following the experimental protocol (H&E staining; ×400)
Effect of Atorvastatin on Myocardial Apoptosis After CME
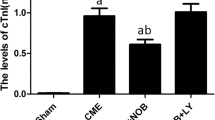
Using TUNEL assay, the dead cells in the myocardial nuclei were stained yellow brown, whereas the normal cells were stained light blue. Myocardial apoptosis after CME was revealed primarily in the myocardial microinfarction foci and the peripheral zones. In sham animals, we occasionally observed myocardial apoptosis occurring in the subendocardium and papillary muscles. The myocardial apoptotic percentages in sham, CME, lavage control, CHO, and atorvastatin groups were 0.20 ± 0.15, 3.17 ± 1.26, 3.03 ± 1.11, 1.01 ± 0.45, and 1.34 ± 0.27, respectively. CME group had a significantly increased (P < 0.05) apoptotic rate as compared to sham group. CHO and atorvastatin groups had significantly decreased (P < 0.05) apoptotic rate as compared to CME group (Figs. 3 and 4).

Graph showing effect of atorvastatin on apoptosis after CME. Myocardial apoptosis after CME (TUNEL assay) was detected primarily in the myocardial microinfarction foci and the peripheral zones. In sham animals, myocardial apoptosis was occasionally found in the subendocardium and papillary muscles. The data (n = 10) are expressed as means ± SD. The number (#) sign represents a P-value <0.05 as compared with sham group, whereas an asterisk (*) represents a P-value <0.05 as compared with CME and lavage control groups

Photomicrographs showing effect of atorvastatin on apoptosis after CME. TUNEL assay reveals the myocardial apoptosis in sham control (#1); CME (#2); lavage control (#3); CHO (#4); and atorvastatin (#5) groups. Normal cell nuclei are stained pale blue, while the apoptotic cardiomyocyte nuclei (arrows) are stained brown (×400) (Color figure online)
Effect of Atorvastatin on Caspase-3 and -8 Activation Levels
Activated caspase-3 and -8 proteins were detected using western blot analysis (Figs. 5 and 6). CME and lavage control groups had significantly increased (P < 0.05) expression of activated caspase-3 and -8 in myocardial cells as compared to sham group. Atorvastatin group showed significantly decreased (P < 0.05) levels of activated caspase-3 and -8 as compared to CME group. CHO treatment specifically inhibited caspase-8 expression (P < 0.05) and decreased caspase-3 levels (P < 0.05). The difference between CHO and CME groups was found to be statistically significant (P < 0.05).

Effect of atorvastatin on caspase-3 expression after CME. a The relative expression levels of activated caspase-3 protein were determined using western blot; the IA values were normalized to GADPH expression levels. Lanes 1, 2, 3, 4, and 5 in the representative gel show caspase-3 expression in sham, CME (after 6 h), lavage control, CHO, and atorvastatin groups, respectively. b The data obtained (n = 10) were expressed and compared between groups as means ± SD values, herein presented graphically. The number (#) sign represents a P-value <0.05 as compared with sham group, whereas an asterisk (*) represents a P-value <0.05 as compared with CME and lavage control groups

Effect of atorvastatin on caspase-8 expression after CME. a The relative expression levels of activated caspase-8 protein were determined using western blot; the IA values were normalized to GADPH expression levels. Lanes 1, 2, 3, 4, and 5 in the representative gel show caspase-8 expression in sham, CME (after 6 h), lavage control, CHO, and atorvastatin groups, respectively. b The data obtained (n = 10) were expressed and compared between groups as means ± SD values, herein presented graphically. The number (#) sign represents a P-value <0.05 as compared with sham group, whereas an asterisk (*) represents a P-value <0.05 as compared with CME and lavage control groups
Discussion
CME is a common complication that occurs following atherosclerotic plaque rupture after ACS and is a bridge to vascular intervention. CME, as an independent predictor of poor long-term prognosis in patients with acute myocardial infarction [3], causes transient “no blood flow” or “slow flow” [1, 2]. Animals’ studies have shown that during the acute phase following CME, local myocardium exhibits microinfarction foci, necrosis and apoptosis of the myocardial cells, and a decline in cardiac function [8]. It was reported [13] that atorvastatin treatment could protect against ischemia–reperfusion injury. Using atorvastatin as an acute short-term therapy at days 1 and 3 produced a protective effect following myocardial ischemia–reperfusion injury and also reduced the area of myocardial infarction [14]. Besides, the risk for perioperative myocardial injury was significantly reduced if the patients with stable angina were administered atorvastatin therapy (40 mg/day) for 7 days before PCI [6].
The potential mechanism(s) of action of atorvastatin during the early stages of protection remain undefined; however, this effect is independent of cholesterol which requires much more time to occur. In addition to the lowering of lipids, atorvastatin plays numerous roles as follows: (1) Early administration of atorvastatin can reduce the inflammatory cell infiltration into cardiac muscle tissue, oxidative stress, and adhesion of monocytes [13]; (2) Atorvastatin can increase the bioavailability of nitric oxide and nitric oxide can reduce the myocardial ischemia–reperfusion injury in vivo; while, atorvastatin therapy can improve the bioavailability of nitric oxide and inhibit cell apoptosis through the Akt pathway; (3) Atorvastatin can reduce the left ventricular remodeling following acute myocardial infarction. It was reported [15] that following ligation of the coronary artery for 6 h, atorvastatin reduced the left ventricular remodeling as well as the activity of matrix metalloproteinases, and also improved contractile dysfunction after 28 days.
During our preliminary studies, we found that microinfarction foci were observed in nearly all sample slices following infusion of 3,000 microembolization balls. Thus, we used this procedure as a rodent model of CME. The peak of myocardial apoptosis appeared 6 h after CME. Since the linear correlation analysis indicated that cardiac function was negatively correlated with myocardial apoptotic rate; we, therefore, chose 6-h post-CME as the observation time point in this study. Notably, our data show that at 6 h post-CME, multiple microinfarction foci appeared in the myocardium, myocardial cells became apoptotic and necrotic, and the cardiac function was decreased.
Caspase activation is considered a key to the induction of apoptosis [16]. Caspase-3 is the key protease in mammalian cell apoptosis, the main apoptotic effect factor and the most important player involved in apoptosis. Evidence from the previous studies [17, 18] suggests that the apoptosis characteristics such as chromosome condensation and DNA fragmentation are present which are directly related to caspase-3. Using immunohistochemical double staining, Krupinski et al. [19] showed that the expression of TUNEL-positive cells in the infarct and infarct border zone were consistent with the caspase-3 expression. Daily atorvastatin therapy for 7 days before surgery significantly improved myocardial apoptosis and systolic dysfunction caused by CME. Molecular biology techniques revealed a significant inhibition of caspase-3 protein activation in the myocardial tissue, a reduction in the expression of activated caspase-8 protein, and a reduction in the myocardial apoptosis rate. Cardiac echocardiography revealed systolic dysfunction and the left ventricular dilation manifested by a decrease in LVEF, FS, and CO as well as an increase in LVEDd. Since myocardial cells are non-differentiating, inhibition of myocardial apoptosis may be one of the most important mechanisms by which atorvastatin therapy improves the prognosis of CME.
Herein, we showed that atorvastatin pretreatment significantly decreased activated caspase-8 protein expression in a rodent model of CME. This inhibitory effect was the same as that achieved using CHO (specific inhibitor of caspase-8), indicating that atorvastatin-related inhibition of myocardial apoptosis is associated with blockade of the death receptor-mediated apoptosis pathway. In addition, we also found that the expression of activated caspase-3 protein after pretreatment with atorvastatin in CME group was lower than that using specific inhibitor of caspase-8 in CHO group. This could be the result of statins having a variety of effects on cardiac apoptosis.
However, some limitations of our study deserve consideration. First, polymer microspheres can lead to mechanical plugging of micro-vessels, but they do not have the biochemical characteristics of natural microemboli including platelets, endothelial cells, and plaque debris with cholesterol crystals. Second, natural emboli exhibit a wide range of sizes and also differ in shape. The microspheres inducing CME used in this study may not entirely reflect the nature of clinical CME. Therefore, the microspheres with more similarities to ruptured plaques will be used in our future studies.
In conclusion, atorvastatin can inhibit the activation of caspase-8, a key apoptotic protein involved in myocardial death receptor-mediated apoptosis pathway and hence prevents the myocardial apoptosis after CME in rats. To our understanding, this may be an important mechanism by which atorvastatin improves the prognosis of CME patients.
References
Kawano, H., Hayashida, T., Ohtani, H., et al. (2005). Histopathological findings of the no-reflow phenomenon following coronary intervention for acute coronary syndrome. International Heart Journal, 46, 327–332.
Jaffe, R., Charron, T., Puley, G., Dick, A., & Strauss, B. H. (2008). Microvascular obstruction and the no-reflow phenomenon after percutaneous coronary intervention. Circulation, 117, 3152–3156.
Itsuro, M., Takahito, S., Kenji, O., et al. (2000). Angiographic no-reflow phenomenon as a predictor of adverse long-term outcome in patients treated with percutaneous transluminal coronary angioplasty for first acute myocardial infarction. Journal of the American College of Cardiology, 36, 1202–1209.
Grube, E., Schofer, J. J., Webb, J., et al. (2002). Evaluation of a balloon occlusion and aspiration system for protection from distal embolization during stenting in saphenous vein grafts. American Journal of Cardiology, 89, 941–945.
Patti, G., Pasceri, V., Colonna, G., et al. (2007). Atorvastatin pretreatment improves outcomes in patients with acute coronary syndromes undergoing early percutaneous coronary intervention: Results of the ARMYDA-ACS randomized trial. Journal of the American College of Cardiology, 49, 1272–1278.
Pasceri, V., Patti, G., Nusca, A., et al. (2004). Randomized trial of atorvastatin for reduction of myocardial damage during coronary intervention: Results from the ARMYDA (atorvastatin for reduction of myocardial damage during angioplasty) Study. Circulation, 110, 674–678.
Scalia, R. (2005). Statins and the response to myocardial injury. American Journal of Cardiovascular Drugs, 5, 163–170.
Thielmann, M., Dörge, H., Martin, C., et al. (2002). Myocardial dysfunction with coronary microembolization: Signal transduction through a sequence of nitric oxide, tumor necrosis factor-alpha and sphingosine. Circulation Research, 90, 807–813.
Kalenikova, E. I., Gorodetskaya, E. A., Zacharova, N. V., Shechter, A. B., & Medvedev, O. S. (1998). Perindopril effects on angiotensin I elimination in lung after experimental myocardial injury induced by intracoronary microembolization in rats. Journal of Cardiovascular Pharmacology, 32, 608–615.
Bajt, M. L., Lawson, J. A., Vonderfecht, S. L., Gujral, J. S., & Jaeschke, H. (2000). Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: Evidence for a postmitochondrial processing of caspase-8. Toxicological Sciences, 58, 109–117.
Schiller, N. B., Acquatella, H., Ports, T. A., et al. (1979). Left ventricular volume from paired biplane two-dimensional echocardiography. Circulation, 60, 547–555.
Zhang, Q. Y., Ge, J. B., Chen, J. Z., et al. (2006). Mast cell contributes to cardiomyocyte apoptosis after coronary microembolization. Journal of Histochemistry and Cytochemistry, 54, 515–523.
Bell, R. M., & Yellon, D. M. (2003). Atorvastatin, administrated at the onset of reperfusion, and independent of lipid lowering, protects the myocardium by regulating a prosurvival pathway. Journal of the American College of Cardiology, 41, 508–515.
Mensah, K., Mocanu, M. M., & Yellon, D. M. (2005). Failure to protect the myocardium against ischemia/reperfusion injury after chronic atorvastatin treatment is recaptured by acute atorvastatin treatment. Journal of the American College of Cardiology, 45, 1287–1291.
Fonarow, G. C., Wright, R. S., Spencer, F. A., et al. (2005). Effect of statin use within the first 24 hours of admission for acute myocardial infarction on early morbidity and mortality. American Journal of Cardiology, 96, 611–616.
Holeyman, C. R., & Larson, D. F. (2001). Apoptosis in the ischemic-reperfused myocardium. Perfusion, 16, 491–502.
Saikumar, P., Dong, Z., Mikhailov, V., Denton, M., Weinberg, J. M., & Venkatachalam, M. A. (1999). Apoptosis: definition, mechanism and relevance to disease. American Journal of Medicine, 107, 489–506.
Porter, A. G., & Janicke, R. U. (1999). Emerging roles of caspase-3 in apoptosis. Cell Death and Differentiation, 6, 99–104.
Krupinski, J., Lopez, E., Marti, E., & Ferrer, I. (2000). Expression of caspases and their substrates in the rat model of focal cerebral ischemia. Neurobiology of Disease, 7, 332–342.
Acknowledgments
We thank the National Natural Science Foundation of China for financial support (Grant No. 30760262/C030313).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, L., Su, Q., Wang, Y. et al. Effect of Atorvastatin (Lipitor) on Myocardial Apoptosis and Caspase-8 Activation Following Coronary Microembolization. Cell Biochem Biophys 61, 399–406 (2011). https://doi.org/10.1007/s12013-011-9199-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-011-9199-z