
Abstract
To explore the mechanism by which rosuvastatin prevents coronary microembolism (CME)-induced cardiac injury and cardiomyocyte apoptosis. Animal and cell models of CME were established and treated with different doses of rosuvastatin. Echocardiography and histological staining were applied to assess left ventricular function and cardiac injury. Masson trichrome staining was used to evaluate fibrin deposition in the myocardium. The activity of lactate dehydrogenase (LDH) in serum and cell culture supernatant was detected. TUNEL staining and flow cytometry were used to evaluate apoptosis in myocardium and cardiomyocytes, respectively. The activity of ROS was revealed by DHE staining. The expression levels of Nox2, cleaved caspase-3, cytochrome C, p53, Bax and Bcl-2 were also detected. Rosuvastatin pretreatment improved the left ventricular function of CME mice and reduced inflammatory cell infiltration and fibrin deposition in the myocardium. Rosuvastatin reduced the production of ROS by inhibiting the expression of Nox2. Rosuvastatin also downregulated pro-apoptotic proteins cleaved caspase-3, cytochrome C, p53 and Bax, and upregulated anti-apoptotic Bcl-2. Rosuvastatin mitigates CME-induced cardiac injury by inhibiting Nox2-induced ROS overproduction and alleviating p53/Bax/Bcl-2-dependent cardiomyocyte apoptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases contribute to a large portion of global deaths, producing considerable health and economic burden [1, 2]. Deaths from ischemic heart disease have significantly increased in the past two decades in China and worldwide [2, 3]. The application of cardiac catheterizations is still tremendous [2]. However, periprocedural myocardial injury or infarction, characterized by an asymptomatic or symptomatic elevation of cardiac biomarker expression, may occur during coronary revascularization surgery [4, 5]. Periprocedural myocardial injury is a common event following transcatheter aortic valve replacement and confers increased risks of both short-term and long-term deaths [6]. Myocardial infarction associated with percutaneous coronary intervention (type 4a) or coronary artery bypass surgery (type 5) can lead to an even worse clinical outcome [7,8,9,10].
Coronary microembolization (CME) is a predominant cause of periprocedural myocardial injury and infarction [11, 12]. Atherosclerotic plaque rupture represents a decisive event in acute myocardial infarction, causing not only occlusive coronary thrombosis but also embolization of atherosclerotic and thrombotic debris into the coronary microcirculation [13]. Atherosclerotic debris and superimposed thrombotic material give rise to patchy microinfarcts accompanied by inflammatory responses in the coronary microcirculation [14]. The clinical frequency and importance of CME have been recognized and appreciated for over two decades [12], but there is no optimal treatment dealing with CME yet.
Rosuvastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, has been widely used for management of ischemic heart diseases due to its efficient cholesterol-lowering function. Many large-scale clinical randomized controlled trials have shown that rosuvastatin treatment can reduce the morbidity and mortality from cardiovascular diseases [15,16,17]. Moreover, short-term rosuvastatin administration can depress the incidence of perioperative myocardial injury as well as other cardiovascular events following coronary intervention [18,19,20,21]. Considering the potent efficacy of rosuvastatin treatments, the biological effects of rosuvastatin beyond regulation of cholesterol are worthy of further exploration.
This study investigates if short-term high-dose rosuvastatin imposes protection against CME since the role of rosuvastatin in the management of CME has been rarely studied. Furthermore, this study deciphers the potential molecular mechanism for the cardioprotective effect of rosuvastatin.
Materials and Methods
Ethical Statement
The use of rat ventricles for isolation of cardiomyocytes and the following animal experiments were authorized by the Animal Care and Use Committee of Fudan University (Shanghai, China), following the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication No. 85-23, revised 1996).
Cell Culture
Ventricles were collected from 1- to 3-day-old Sprague–Dawley rats (the Department of Laboratory Animal Science, Fudan University, Shanghai, China) and trypsinized with 0.1% trypsin (Gibco, Grand Island, NY, USA) for 2 h. Primary cardiomyocytes were separated from the trypsinized ventricles and cultured in low glucose Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco), 10 µg/mL streptomycin (HyClone Laboratories Inc., Logan, UT, USA) and 10 U/mL penicillin (HyClone Laboratories Inc.) following the standard protocols. The culture medium was replaced by antibiotics-free DMEM (containing 10% FBS) after 24 h. The isolated cardiomyocytes were divided into six groups: control group, TNF-α group, 10 μmol/L rosuvastatin group, 20 μmol/L rosuvastatin group, 10 μmol/L rosuvastatin + TNF-α group, and 20 μmol/L rosuvastatin + TNF-α group. Cardiomyocytes in the control group were cultured under normal conditions without any additional treatment, while cells in the TNF-α group were incubated with TNF-α (40 ng/mL, PeproTech Inc., Rocky Hill, NJ, USA) for 48 h. Cardiomyocytes in the 10 μmol/L rosuvastatin and 20 μmol/L rosuvastatin groups were respectively treated with 10 μmol/L or 20 μmol/L rosuvastatin (AstraZeneca, London, UK) for 54 h. Cardiomyocytes in the 10 μmol/L rosuvastatin + TNF-α group and 20 μmol/L rosuvastatin + TNF-α group were pretreated with 10 μmol/L or 20 μmol/L rosuvastatin for 6 h before TNF-α treatment (40 ng/mL, 48 h).
Animal Experiments
Twenty-four male C57BL/6 mice (20–25 g, the Department of Laboratory Animal Science, Fudan University) were randomized into 4 groups (6 mice per group): sham group, CME group, 10 mg/kg rosuvastatin + CME group, and 20 mg/kg rosuvastatin + CME group. Rosuvastatin (10 or 20 mg/kg) was orally administered from 7 days before operation to 3 days post-op. Purified water instead of rosuvastatin was orally given to the sham and CME groups. On day 3 post-op, an echocardiography assessment was performed, venous blood samples were collected, and the mice were sacrificed by cervical dislocation for collection of heart tissues after inhalation 99.99% carbon dioxide (CO2) for 5 min.
CME Model Establishment
Detailed experimental procedures for generating a CME model have been described in a previous study [22]. Mice were fully anesthetized by inhalation of 2% isoflurane (Baxter International Inc., Deerfield, IL, USA), and the anesthesia of mice was sustained by 1.5% isoflurane during the following procedures. A small animal ventilator (Type 7025, Ugo Basile, Comerio, Italy) was used to assist the respiration of the mice (100 breaths per minute) during the exposure of heart and ascending aorta. The ascending aorta was occluded for 15 s, during which a bolus (5 × 105 microspheres) of white-stained polystyrene microspheres (9 μm, 50 μL, Dynospheres, Dyno Particles, Lillestrøm, Norway) was injected with a 29-gauge needle into the left ventricle. The sham group underwent identical procedures during which microspheres were replaced by saline.
Echocardiography
Cardiac function assessment was performed before operation and 3 days post-op using a high resolution small animal ultrasonic imaging system (Vevo 2100, Visual Sonics, Toronto, Canada). According to the manual of Vevo 2100, mice were fully anesthetized by inhalation of 1–2% isoflurane in oxygen and placed on a temperature-controlled (37 °C) plate. The limbs of the mice were fixed and connected to 4 electrodes. The concentration of isoflurane was regulated to keep the heart rates of the mice at 500 bpm. Left ventricular end-diastolic diameter (LVEDD), end-systolic diameter (LVESD), ejection fraction (LVEF) and fractional shortening (LVFS) were automatically calculated by Vevo 2100.
Hematoxylin–Eosin (H&E) Staining and Masson Trichrome Staining
After fixation with 10% formaldehyde, heart tissues were paraffin-embedded and sectioned into 5-μm slices for histological staining. The stained slices were evaluated by Olympus BX-51 light microscopy (Olympus, Tokyo, Japan). For each mouse, an average of three slices from diverse areas of the heart was obtained using Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA). The degree of inflammatory cell infiltration/fibrin deposition in a slice was presented as the ratio of infiltration/deposition area to total area.
Evaluation of Myocardial Damage
Serum was separated from the blood samples collected from postocular veins. The activity of lactate dehydrogenase (LDH) in mouse serum or cell culture supernatant was determined by a LDH cytotoxicity assay detection kit (R&D Systems, Minneapolis, MN, USA) based on the instructions. The optical density (450 nm) was measured by a microplate reader (Bio-rad 550, Bio-Rad Laboratories, Hercules, CA, USA).
Evaluation of Reactive Oxygen Species (ROS) Production
Frozen myocardium sections or cardiomyocytes were washed with preheated PBS (37 °C). The tissues were stained with 10 μmol/L dihydroethidium (DHE) (dissolved in PBS; Sigma Aldrich, St. Louis, MO, USA) and the cells were stained with 5 µM DHE (dissolved in FBS-free DMEM) for 30 min (37 °C, away from light). After staining, the sections were washed three times with PBS, 3 to 5 min each time. For each mouse, an average fluorescence intensity of three slices from diverse areas of the heart was obtained using Image-Pro Plus 6.0. The fluorescence intensity in the cells was detected by a FACScan™ flow cytometer (Becton Dickson, Franklin Lakes, NJ, USA).
TUNEL Assay
Frozen myocardium sections were washed, fixed with 4% formaldehyde in PBS for 20 min, and permeabilized with 0.1% Triton X-100 for 2 min before the blinded in situ detection of apoptosis (Roche Diagnostics, Shanghai, China) and DAPI staining (Beyotime, Shanghai, China). For each mouse, three slices from different areas of the heart were selected and the results were averaged. The apoptotic index (AI) = TUNEL-positive cells (green fluorescent)/total cells (blue fluorescent).
Western Blotting
Proteins separated by SDS-PAGE were transferred onto PVDF membranes (Millipore, Belford, MA, USA). The membranes were incubated with 5% bovine serum albumin in Tris-buffered saline containing 0.1% Tween 20, and then with primary rabbit-anti human antibodies against NADPH oxidase 2 (Nox2, ab129068, 1:5000, Abcam, Massachusetts, USA), p53 (2527, 1:1000), Bax (5023, 1:1000), Bcl-2 (3498, 1:1000), cytochrome C (11,940, 1:1000), cleaved caspase-3 (9661, 1:1000) and GAPDH (5174, 1:1000) (Cell Signaling Technology, Beverly, MA, USA). The blots were developed using HRP-conjugated secondary goat-anti rabbit antibodies (Abbkine, Redlands, CA, USA) and SuperSignal West Pico chemiluminescent substrate (Pierce Biotechnology, Rockford, IL, USA), and quantitated by Image-Pro Plus 6.0.
Immunohistochemistry
Deparaffinized heart tissue sections were permeabilized, blocked at room temperature, and incubated with Nox2 and cleaved caspase-3 antibodies (Cell Signaling Technology, Beverly, MA, USA) at 4 °C overnight, followed by incubation with HRP-labeled secondary antibodies (Abbkine, Redlands, CA, USA). Finally, the sections were treated with DAB using HRP Color Development Kit (Beyotime, Shanghai, China).
Flow Cytometry
Trypsinized cardiomyocytes were washed in 37 °C PBS and stained with Annexin V and propidium iodide (PI) (eBioscience, San Diego, CA, USA). Apoptotic cardiomyocytes were sought out by fluorescence-labeled Annexin V on the FACScan™ flow cytometer (Becton Dickson, Franklin Lakes, NJ, USA). The percentages of Annexin V-positive cells and PI-negative cells were calculated. The data were converted into dot plots by Cell Quest Software (Becton Dickinson).
Statistical Analysis
Statistical analyses were performed using Stata 10.0 (StataCorp, TX, USA). Groups were compared by one-way analysis of variance, and a Bonferroni’s test was performed to identify significant differences among groups. All data were presented as mean ± standard deviation. P values were two-sided, and P < 0.05 indicated a statistically significant difference.
Results
Rosuvastatin Improves Left Ventricular Function Following CME
CME mouse models were established and treated with 10 or 20 mg/kg rosuvastatin. The echocardiography assessment on day 3 post-op showed that LVEF and LVFS were significantly decreased in the CME group while restored in the rosuvastatin treatment groups (Table 1) (LVEF: PCME vs sham = 0.027, P10 mg/kg rosuvastatin + CME vs CME = 0.042, P20 mg/kg rosuvastatin + CME vs CME = 0.046; LVFS: PCME vs sham = 0.001, P10 mg/kg rosuvastatin + CME vs CME = 0.003, P20 mg/kg rosuvastatin + CME vs CME = 0.012). LVESD and LVEDD were enlarged in the CME group while narrowed in both the rosuvastatin treatment groups (Table 1) (LVESD: PCME vs sham = 0.030, P10 mg/kg rosuvastatin + CME vs CME = 0.013, P20 mg/kg rosuvastatin + CME vs CME = 0.034; LVEDD: PCME vs sham = 0.028, P10 mg/kg rosuvastatin + CME vs CME = 0.036, P20 mg/kg rosuvastatin + CME vs CME = 0.025).
Rosuvastatin Mitigates CME-Induced Cardiac Injury
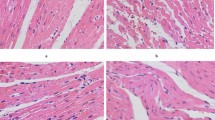
The model mice were treated with different doses of rosuvastatin. The histopathological evaluation of myocardium was carried out 3 days post-op using H&E staining and Masson staining. The results of H&E staining showed obvious inflammatory cell infiltration in the CME group (7.42 ± 1.23%) in comparison with the sham group (0.57 ± 0.05%) (Fig. 1A, B, P < 0.001). The inflammatory cell infiltration was significantly alleviated in both the 10 mg/kg rosuvastatin group (3.34 ± 0.45%) and 20 mg/kg rosuvastatin group (4.33 ± 0.39%) (Fig. 1A, B, P10 mg/kg rosuvastatin + CME vs CME = 0.005, P20 mg/kg rosuvastatin + CME vs CME = 0.004). The results of Masson staining indicated distinct fibrous deposition in the microinfarcts of the CME group (6.89 ± 0.77%) compared to the sham group (0.44 ± 0.06%) (Fig. 1A, C, P < 0.001). High-dose rosuvastatin treatment showed a more significant effect on reducing fibrous deposition (Fig. 1A, C, P10 mg/kg rosuvastatin + CME vs CME = 0.012, P20 mg/kg rosuvastatin + CME vs CME = 0.008). Moreover, the serum level of LDH was elevated in the CME group and reduced in the rosuvastatin treatment groups (Fig. 1D, PCME vs sham < 0.001, P10 mg/kg rosuvastatin + CME vs CME < 0.01, P20 mg/kg rosuvastatin + CME vs CME < 0.01).

Rosuvastatin mitigates CME-induced cardiac injury. Note CME model mice were treated with 10 or 20 mg/kg rosuvastatin from 7 days before operation to 3 days post-op. The heart tissues and serum were collected 3 days after operation. A The representative images (× 25) of H&E staining and Masson trichrome staining; the extent of inflammatory cell infiltration (B) and fibrin deposition C in the myocardium; D the activity of serum LDH. ***P < 0.001, vs the sham group; #P < 0.05, ##P < 0.01, §§P < 0.01, vs the CME group. CME coronary microembolization; LDH lactate dehydrogenase
Rosuvastatin Reduces Nox2 Expression and ROS Production in CME Mice
The myocardium tissues were collected 3 days after CME modeling. DHE staining revealed that the CME group had a higher level of ROS production than the sham group (Fig. 2A, B, P < 0.001). Rosuvastatin intervention significantly decreased the production of ROS (Fig. 2A, B, P10 mg/kg rosuvastatin + CME vs CME < 0.001, P20 mg/kg rosuvastatin + CME vs CME < 0.001). The results of immunohistochemistry and western blotting both demonstrated that the CME group had a higher expression of Nox2 protein than the sham group (Fig. 2C, D, P = 0.032). Nox2 was downregulated in the rosuvastatin treatment groups (Fig. 2C, D, P10 mg/kg rosuvastatin + CME vs CME = 0.041, P20 mg/kg rosuvastatin + CME vs CME = 0.009). Taken together, rosuvastatin reduced the production of ROS as well as the expression of Nox2 in the myocardium following CME.

Rosuvastatin reduces Nox2 expression and ROS production in CME mice. Note CME model mice were treated with 10 or 20 mg/kg rosuvastatin from 7 days before operation to 3 days post-op. The myocardium tissues were collected 3 days after operation. A The representative images (× 200) of DHE staining; B the fluorescence intensity of ROS in each group; C the representative images (× 200) of immunohistochemistry for visualizing the expression of Nox2 protein; D western blotting analysis of the expression of Nox2 protein. *P < 0.05, ***P < 0.001, vs the sham group; #P < 0.05, ###P < 0.001, §§ P < 0.01, §§§P < 0.001, vs the CME group. ROS reactive oxygen species; CME coronary microembolization; DHE dihydroethidium
Rosuvastatin Inhibits Myocardial Apoptosis via p53/Bax/Bcl-2 Signaling Pathway in CME Mice
The myocardium tissues were collected for apoptosis analysis on day 3 post-op. The CME group showed more TUNEL-positive cells than the sham group (Fig. 3A, P < 0.001). Rosuvastatin treatment significantly reduced CME-induced myocardial apoptosis (Fig. 3A, P10 mg/kg rosuvastatin + CME vs CME < 0.001, P20 mg/kg rosuvastatin + CME vs CME < 0.001). The expression of cleaved caspase-3 was higher in the CME group than in the sham group (Fig. 3B, C, P = 0.005). A decline in the expression of cleaved caspase-3 was found in the rosuvastatin treatment groups (Fig. 3B, C, P10 mg/kg rosuvastatin + CME vs CME = 0.038, P20 mg/kg rosuvastatin + CME vs CME = 0.007).

Rosuvastatin inhibits myocardial apoptosis via p53/Bax/Bcl-2 signaling pathway in CME mice. Note CME model mice were treated with 10 or 20 mg/kg rosuvastatin from 7 days before operation to 3 days post-op. The myocardium tissues were collected 3 days after operation. A The representative images (× 400) of TUNEL assay for detecting myocardial apoptosis; B the representative images (× 200) of immunohistochemistry for visualizing the expression of cleaved caspase-3; western blotting analysis of the expression of cleaved caspase-3 (C), cytochrome-C (D), p53 (E), Bax (F) and Bcl-2 (G). *P < 0.05, **P < 0.01, ***P < 0.001, vs the sham group; #P < 0.05, ##P < 0.01, ###P < 0.001, §P < 0.05, §§P < 0.01, §§§P < 0.001, vs the CME group. CME coronary microembolization
The western blotting further showed that the release of cytochrome C was augmented in the CME group compared with the sham group (Fig. 3D, P = 0.004). The expression of cytochrome C was decreased after rosuvastatin treatment (Fig. 3D, P10 mg/kg rosuvastatin + CME vs CME = 0.007, P20 mg/kg rosuvastatin + CME vs CME = 0.047). The expression of p53 and Bax proteins was increased in the CME group (Fig. 3E, F, Pp53 = 0.021, PBax = 0.035, vs the sham group), and decreased in the 10 mg/kg rosuvastatin + CME group (Fig. 3E, F, Pp53 = 0.047, PBax = 0.036, vs the CME group) and 20 mg/kg rosuvastatin + CME group (Fig. 3E, F, Pp53 = 0.042, PBax = 0.026, vs the CME group). Moreover, Bcl-2 was downregulated in the CME group (Fig. 3G, P = 0.043, vs the sham group) and upregulated after rosuvastatin treatment (Fig. 3G, P10 mg/kg rosuvastatin + CME vs CME = 0.019, P20 mg/kg rosuvastatin + CME vs CME = 0.005). The above data indicated that rosuvastatin inhibited CME-induced myocardial apoptosis by regulating the p53/Bax/Bcl-2 signaling pathway.
Rosuvastatin Alleviates TNF-α-induced Cardiomyocyte Apoptosis Via p53/Bax/Bcl-2 Signaling Pathway
TNF-α was used to induce apoptosis in cardiomyocytes. The cells were pretreated with different doses of rosuvastatin before TNF-α treatment. The viability of cardiomyocytes was decreased in the TNF-α group (Fig. 4A, P < 0.001, vs the control group) and increased in the rosuvastatin + TNF-α treatment groups (Fig. 4A, P10 μM rosuvastatin + TNF-α vs TNF-α < 0.05, P20 μM rosuvastatin + TNF-α vs TNF-α < 0.01). A marked elevation of LDH activity was detected in the TNF-α group (Fig. 4B, P < 0.001, vs the control group). The release of LDH was decreased in the rosuvastatin + TNF-α treatment groups (Fig. 4B, P10 μM rosuvastatin + TNF-α vs TNF-α < 0.05, P20 μM rosuvastatin + TNF-α vs TNF-α < 0.01). The TNF-α group also showed a higher apoptosis rate than the control group (Fig. 4C, P < 0.001). Rosuvastatin treatment reduced TNF-α-induced cardiomyocyte apoptosis (Fig. 4C, P10 μM rosuvastatin + TNF-α vs TNF-α < 0.05, P 20 μM rosuvastatin + TNF-α vs TNF-α < 0.01).

Rosuvastatin alleviates TNF-α-induced cardiomyocyte apoptosis via p53/Bax/Bcl-2 signaling pathway. Note Cardiomyocytes were pretreated with different doses of rosuvastatin before TNF-α treatment. A CCK-8 assay for assessing cell viability; B the activity of lactate dehydrogenase in the cell culture supernatant; C flow cytometry for evaluating cell apoptosis; D flow cytometry for detecting dihydroethidium florescence intensity; E western blotting analysis of the expression of Nox2; F western blotting analysis of the expression of cleaved caspase-3 (G), cytochrome-C (H), p53 (I), Bax (J) and Bcl-2 (K). **P < 0.01, ***P < 0.001, vs the control group; #P < 0.05, ##P < 0.01, vs the TNF-α group
The intensity of DHE fluorescence detected by flow cytometry indicated that the production of ROS was increased in the TNF-α group (Fig. 4D, P < 0.001, vs the control group) and decreased in the rosuvastatin + TNF-α treatment groups (Fig. 4D, P10 μM rosuvastatin + TNF-α vs TNF-α < 0.05, P20 μM rosuvastatin + TNF-α vs TNF-α < 0.01). The western blotting showed that Nox2 was upregulated in the TNF-α group (Fig. 4E, P < 0.001, vs the control group) and downregulated in the rosuvastatin + TNF-α treatment groups (Fig. 4E, P10 μM rosuvastatin + TNF-α vs TNF-α < 0.01, P20 μM rosuvastatin + TNF-α vs TNF-α < 0.01). Moreover, increases in the expression of cleaved caspase-3 (P < 0.001), cytochrome-C (P < 0.01), p53 (P < 0.01) and Bax (P < 0.001) and a decrease in the expression of Bcl-2 (P < 0.001) were found in the TNF-α group (Fig. 4F–K, vs the control group). The expression trends of these proteins were reversed in the rosuvastatin + TNF-α treatment groups (Fig. 4F–K, P < 0.05).
Rosuvastatin treatment alone did not affect the cellular activity and protein expression in cardiomyocytes. Rosuvastatin alleviated TNF-α-induced cardiomyocyte apoptosis by regulating the p53/Bax/Bcl-2 signaling pathway.
Discussion
Perioperative myocardial injury, manifested by chest pain or discomfort and an increase in postoperative cardiac troponin, can be brought about by coronary intervention-induced CME. In this study, we established animal and cell models of CME to explore the molecular mechanism for CME-induced injury. The cardiac injury indicator LDH, focal inflammatory cell infiltration and fibrin deposition were increased while the cardiac contractile function was impaired at the early stage of CME. Rosuvastatin improved cardiac function and reduced cardiomyocyte apoptosis following CME.
Rosuvastatin is an efficient blood lipid-regulating drug that can be used to prevent coronary heart disease. Long-term use of rosuvastatin reduces coronary atherosclerotic plaque burden and lipid accumulation [23, 24], and can even reverse coronary atherosclerotic plaque [25]. Preoperative high-dose rosuvastatin therapy significantly prevented perioperative myocardial infarction following elective coronary intervention [7, 26]. Rosuvastatin preloading could also reduce the infarct size and ischemia–reperfusion injury following coronary intervention [27] as well as decreasing cardiovascular events and short-term mortality [20, 28]. In addition to anti-cholesterol effects, rosuvastatin was also been reported for its pleiotropic effects on cardiovascular diseases. For example, rosuvastatin reversed myocardial remodeling in metabolic syndrome through AMPK-mediated antifibrotic effects [29] and reduced myocardial apoptosis and fibrosis through regulating PI3K/Ak and MEK/ERK pathways [30]. In cardiomyopathy rat model, rosuvastatin could alleviate systemic inflammation [31]. In addition, the protective effect of rosuvastatin against CME-induced myocardial injury was found through inhibiting ROS generation, NLRP3 inflammasome activation, and cardiac pyroptosis [32]. Previous study showed that part of the potential mechanism of rosuvastatin against myocardial injury was the inhibition of isoprenoid synthesis, which consequently led to the inhibition of intracellular signaling molecules Rho, Rac and their downstream target Rho kinase (ROCK) [33]. Besides, rosuvastatin can also activate peroxisome proliferator-activated receptor alpha to explain their antiatherogenic actions [34]. The data from previous clinical studies suggested that short-term intervention with high-dose rosuvastatin may reduce perioperative myocardial injury. In the present study, short-term use of either low-dose or high-dose rosuvastatin significantly attenuated CME-induced myocardial injury and retained myocardial contractile function. Rosuvastatin also inhibited focal inflammatory cell infiltration and fibrin deposition in the myocardium.
Inflammatory response is closely associated with oxidative stress in the occurrence and development of various diseases. Excessive oxidative stress impairs the diastolic function of vascular endothelium and induces apoptosis of endothelial cells and inflammatory responses [35, 36]. Dysregulated accumulation of ROS may lead to ischemic cardiomyopathy and cardiac insufficiency [37]. TNF-α has been shown to play a causal role in abnormal cardiac function following CME [38]. Upregulation of TNF-α promotes myocardial oxidative stress, mitochondrial dysfunction and DNA damage [39,40,41]. An overproduction of ROS was observed in CME mice and TNF-α-treated cardiomyocytes in this study. High-dose rosuvastatin reduced CME-stimulated production of ROS, and therefore alleviated the infiltration of inflammatory cells as well as fibrin deposition. Based on the above evidences, short-term myocardial damage and cardiac dysfunction following CME are related to excessive oxidative stress.
In cardiovascular tissues, ROS are mainly produced by the family of NADPH oxidases (Nox) [42]. The Nox family contains seven members, including Nox1-5 and dual oxidase 1–2 (Duox1–2). Myocardial tissue mostly expresses Nox2 and Nox4 which play important roles in myocardial injury and remodeling [42]. Here we found the expression of Nox2 was increased in CME mice and TNF-α-treated cardiomyocytes. Short-term administration of rosuvastatin inhibited CME-stimulated production of ROS by repressing the expression of Nox2.
Cardiomyocyte apoptosis has been demonstrated to be implicated in the impairment of cardiac function following CME in a swine model [43]. The phenomenon of myocardial apoptosis was also found in CME mice, which was reversed by rosuvastatin intervention. p53 is a tumor suppressor gene that encodes a 53 kDa protein. Protein p53 can upregulate the expression of Bax or downregulate the expression of Bcl-2 to promote apoptosis. Bax and Bcl-2 proteins belong to the Bcl-2 family but function differently. The Bcl-2 family is divided into two main subgroups: the pro-apoptotic and the anti-apoptotic. Bax protein alters mitochondrial membrane permeability by forming dimers or combining with Bak protein, leading to the release of cytochrome C and initiation of apoptotic processes [44,45,46,47,48]. On the contrary, Bcl-2 protein antagonizes pro-apoptotic proteins such as Bax. In the present study, p53 and Bax proteins were upregulated and Bcl-2 was downregulated following CME, accompanied by increased release of cytochrome C and caspase-3 protein. These protein expression variations were diminished by rosuvastatin intervention.
Conclusions
In summary, rosuvastatin decreases CME-induced cardiomyocyte apoptosis by regulating the p53/Bax/Bcl-2 signaling pathway, thereby reducing CME-induced cardiac injury and preserving myocardial function. This study sheds light on the biological effects of rosuvastatin other than lipid regulation and provides theoretical basis for the application of rosuvastatin in the management of CME.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Balakumar, P., Maung, U. K., & Jagadeesh, G. (2016). Prevalence and prevention of cardiovascular disease and diabetes mellitus. Pharmacological Research, 113, 600–609.
Writing Group, M., Mozaffarian, D., Benjamin, E. J., Go, A. S., Arnett, D. K., Blaha, M. J., et al. (2016). Heart disease and stroke statistics-2016 update: A report from the American Heart Association. Circulation, 133, e38-360.
Yang, G., Wang, Y., Zeng, Y., Gao, G. F., Liang, X., Zhou, M., et al. (2013). Rapid health transition in China, 1990–2010: Findings from the Global Burden of Disease Study 2010. The Lancet, 381, 1987–2015.
Thygesen, K., Alpert, J. S., Jaffe, A. S., Chaitman, B. R., Bax, J. J., Morrow, D. A., et al. (2018). Fourth universal definition of myocardial infarction. Journal of the American College of Cardiology, 72, 2231–2264.
Zeitouni, M., Silvain, J., Guedeney, P., Kerneis, M., Yan, Y., Overtchouk, P., et al. (2018). Periprocedural myocardial infarction and injury in elective coronary stenting. European Heart Journal, 39, 1100–1109.
Michail, M., Cameron, J. N., Nerlekar, N., Ihdayhid, A. R., McCormick, L. M., Gooley, R., et al. (2018). Periprocedural myocardial injury predicts short- and long-term mortality in patients undergoing transcatheter aortic valve replacement. Circulation. Cardiovascular Interventions, 11, e007106.
Bonaca, M. P., Wiviott, S. D., Braunwald, E., Murphy, S. A., Ruff, C. T., Antman, E. M., et al. (2012). American College of Cardiology/American Heart Association/European Society of Cardiology/World Heart Federation universal definition of myocardial infarction classification system and the risk of cardiovascular death: Observations from the TRITON-TIMI 38 trial (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction 38). Circulation, 125, 577–583.
Davies, W. R., Brown, A. J., Watson, W., McCormick, L. M., West, N. E., Dutka, D. P., et al. (2013). Remote ischemic preconditioning improves outcome at 6 years after elective percutaneous coronary intervention: The CRISP stent trial long-term follow-up. Circulation. Cardiovascular Interventions, 6, 246–251.
Erbel, R., & Wijns, W. (2014). The year in cardiology 2013: Coronary intervention. European Heart Journal, 35, 313–320.
Testa, L., Van Gaal, W. J., Biondi Zoccai, G. G., Agostoni, P., Latini, R. A., Bedogni, F., et al. (2009). Myocardial infarction after percutaneous coronary intervention: A meta-analysis of troponin elevation applying the new universal definition. QJM, 102, 369–378.
Herrmann, J. (2005). Peri-procedural myocardial injury: 2005 update. European Heart Journal, 26, 2493–2519.
Heusch, G., Skyschally, A., & Kleinbongard, P. (2018). Coronary microembolization and microvascular dysfunction. International Journal of Cardiology, 258, 17–23.
Skyschally, A., Leineweber, K., Gres, P., Haude, M., Erbel, R., & Heusch, G. (2006). Coronary microembolization. Basic Research in Cardiology, 101, 373–382.
Heusch, G. (2016). The coronary circulation as a target of cardioprotection. Circulation Research, 118, 1643–1658.
Scandinavian Simvastatin Survival Study Group. (1994). Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). The Lancet, 344, 1383–1389.
LaRosa, J. C., Grundy, S. M., Waters, D. D., Shear, C., Barter, P., Fruchart, J. C., et al. (2005). Intensive lipid lowering with atorvastatin in patients with stable coronary disease. New England Journal of Medicine, 352, 1425–1435.
Long-Term Intervention with Pravastatin in Ischaemic Disease Study G. (1998). Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. New England Journal of Medicine, 339, 1349–1357.
Briguori, C., Madonna, R., Zimarino, M., Calabro, P., Quintavalle, C., Salomone, M., et al. (2016). Rosuvastatin for reduction of myocardial damage during coronary angioplasty—The remedy trial. Cardiovascular Drugs and Therapy, 30, 465–472.
Cay, S., Cagirci, G., Sen, N., Balbay, Y., Durmaz, T., & Aydogdu, S. (2010). Prevention of peri-procedural myocardial injury using a single high loading dose of rosuvastatin. Cardiovascular Drugs and Therapy, 24, 41–47.
Pan, Y., Tan, Y., Li, B., & Li, X. (2015). Efficacy of high-dose rosuvastatin preloading in patients undergoing percutaneous coronary intervention: A meta-analysis of fourteen randomized controlled trials. Lipids in Health and Disease, 14, 97.
Patti, G., Cannon, C. P., Murphy, S. A., Mega, S., Pasceri, V., Briguori, C., et al. (2011). Clinical benefit of statin pretreatment in patients undergoing percutaneous coronary intervention: A collaborative patient-level meta-analysis of 13 randomized studies. Circulation, 123, 1622–1632.
Cao, Y. Y., Chen, Z. W., Jia, J. G., Chen, A., Zhou, Y., Ye, Y., et al. (2016). Establishment of a novel mouse model of coronary microembolization. Chinese Medical Journal (Engl), 129, 2951–2957.
Otagiri, K., Tsutsui, H., Kumazaki, S., Miyashita, Y., Aizawa, K., Koshikawa, M., et al. (2011). Early intervention with rosuvastatin decreases the lipid components of the plaque in acute coronary syndrome: Analysis using integrated backscatter IVUS (ELAN study). Circulation Journal, 75, 633–641.
Raber, L., Taniwaki, M., Zaugg, S., Kelbaek, H., Roffi, M., Holmvang, L., et al. (2015). Effect of high-intensity statin therapy on atherosclerosis in non-infarct-related coronary arteries (IBIS-4): A serial intravascular ultrasonography study. European Heart Journal, 36, 490–500.
Qian, C., Wei, B., Ding, J., Wu, H., Cai, X., Li, B., et al. (2015). Meta-analysis comparing the effects of rosuvastatin versus atorvastatin on regression of coronary atherosclerotic plaques. American Journal of Cardiology, 116, 1521–1526.
Takano, H., Ohba, T., Yamamoto, E., Miyachi, H., Inui, K., Kawanaka, H., et al. (2013). Usefulness of rosuvastatin to prevent periprocedural myocardial injury in patients undergoing elective coronary intervention. American Journal of Cardiology, 111, 1688–1693.
Kim, J. W., Yun, K. H., Kim, E. K., Kim, Y. C., Joe, D. Y., Ko, J. S., et al. (2014). Effect of high dose rosuvastatin loading before primary percutaneous coronary intervention on infarct size in patients with ST-segment elevation myocardial infarction. Korean Circ J, 44, 76–81.
Tentzeris, I., Rohla, M., Jarai, R., Farhan, S., Freynhofer, M. K., Unger, G., et al. (2014). Influence of high-dose highly efficient statins on short-term mortality in patients undergoing percutaneous coronary intervention with stenting for acute coronary syndromes. American Journal of Cardiology, 113, 1099–1104.
Hermida, N., Markl, A., Hamelet, J., Van Assche, T., Vanderper, A., Herijgers, P., et al. (2013). HMGCoA reductase inhibition reverses myocardial fibrosis and diastolic dysfunction through AMP-activated protein kinase activation in a mouse model of metabolic syndrome. Cardiovascular Research, 99, 44–54.
Zhang, Z., Li, S., Cui, M., Gao, X., Sun, D., Qin, X., et al. (2013). Rosuvastatin enhances the therapeutic efficacy of adipose-derived mesenchymal stem cells for myocardial infarction via PI3K/Akt and MEK/ERK pathways. Basic Research in Cardiology, 108, 333.
Cho, D. H., Lim, I. R., Kim, J. H., Kim, M. N., Kim, Y. H., Park, K. H., et al. (2020). Protective effects of statin and angiotensin receptor blocker in a rat model of doxorubicin- and trastuzumab-induced cardiomyopathy. Journal of the American Society of Echocardiography, 33, 1253–1263.
Chen, A., Chen, Z., Zhou, Y., Wu, Y., Xia, Y., Lu, D., et al. (2021). Rosuvastatin protects against coronary microembolization-induced cardiac injury via inhibiting NLRP3 inflammasome activation. Cell Death & Disease, 12, 78.
Zhou, Q., & Liao, J. K. (2010). Pleiotropic effects of statins. Basic research and clinical perspectives. Circulation Journal, 74, 818–826.
Yano, M., Matsumura, T., Senokuchi, T., Ishii, N., Murata, Y., Taketa, K., et al. (2007). Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circulation Research, 100, 1442–1451.
Edirisinghe, I., Yang, S. R., Yao, H., Rajendrasozhan, S., Caito, S., Adenuga, D., et al. (2008). VEGFR-2 inhibition augments cigarette smoke-induced oxidative stress and inflammatory responses leading to endothelial dysfunction. The FASEB Journal, 22, 2297–2310.
Maugeri, N., Rovere-Querini, P., Baldini, M., Baldissera, E., Sabbadini, M. G., Bianchi, M. E., et al. (2014). Oxidative stress elicits platelet/leukocyte inflammatory interactions via HMGB1: A candidate for microvessel injury in sytemic sclerosis. Antioxidants & Redox Signaling, 20, 1060–1074.
Matsushima, S., Tsutsui, H., & Sadoshima, J. (2014). Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends in Cardiovascular Medicine, 24, 202–205.
Dorge, H., Schulz, R., Belosjorow, S., Post, H., van de Sand, A., Konietzka, I., et al. (2002). Coronary microembolization: The role of TNF-alpha in contractile dysfunction. Journal of Molecular and Cellular Cardiology, 34, 51–62.
Cao, Y. Y., Chen, Z. W., Gao, Y. H., Wang, X. X., Ma, J. Y., Chang, S. F., et al. (2015). Exenatide reduces tumor necrosis factor-alpha-induced apoptosis in cardiomyocytes by alleviating mitochondrial dysfunction. Chinese Medical Journal (Engl), 128, 3211–3218.
Mariappan, N., Soorappan, R. N., Haque, M., Sriramula, S., & Francis, J. (2007). TNF-alpha-induced mitochondrial oxidative stress and cardiac dysfunction: Restoration by superoxide dismutase mimetic Tempol. American Journal of Physiology. Heart and Circulatory Physiology, 293, H2726-2737.
Moe, G. W., Marin-Garcia, J., Konig, A., Goldenthal, M., Lu, X., & Feng, Q. (2004). In vivo TNF-alpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. American Journal of Physiology. Heart and Circulatory Physiology, 287, H1813-1820.
Maejima, Y., Kuroda, J., Matsushima, S., Ago, T., & Sadoshima, J. (2011). Regulation of myocardial growth and death by NADPH oxidase. Journal of Molecular and Cellular Cardiology, 50, 408–416.
Chen, Z. W., Qian, J. Y., Ma, J. Y., Chang, S. F., Yun, H., Jin, H., et al. (2014). TNF-alpha-induced cardiomyocyte apoptosis contributes to cardiac dysfunction after coronary microembolization in mini-pigs. Journal of Cellular and Molecular Medicine, 18, 1953–1963.
Antignani, A., & Youle, R. J. (2006). How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Current Opinion in Cell Biology, 18, 685–689.
D’Orsi, B., Mateyka, J., & Prehn, J. H. M. (2017). Control of mitochondrial physiology and cell death by the Bcl-2 family proteins Bax and Bok. Neurochemistry International, 109, 162–170.
Kroemer, G., & Reed, J. C. (2000). Mitochondrial control of cell death. Nature Medicine, 6, 513–519.
Scorrano, L., & Korsmeyer, S. J. (2003). Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochemical and Biophysical Research Communications, 304, 437–444.
Vakifahmetoglu-Norberg, H., Ouchida, A. T., & Norberg, E. (2017). The role of mitochondria in metabolism and cell death. Biochemical and Biophysical Research Communications, 482, 426–431.
Funding
This work was supported by National Natural Science Foundation of China (Grant Nos. 81970295, 81870267, 81800276); Natural Science Foundation of Hunan Province, China (Grant No. 2018JJ3760); Grant of Shanghai Municipal Commission of Health and Family Planning (Grant No. 2017YQ057).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Ethical Approval
The use of rat ventricles for isolation of cardiomyocytes and the following animal experiments were authorized by the Animal Care and Use Committee of Fudan University (Shanghai, China), following the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication No. 85-23, revised 1996).
Informed Consent
Not applicable.
Additional information
Handling Editor: Vittorio Fineschi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cao, Y., Chen, Z., Jia, J. et al. Rosuvastatin Alleviates Coronary Microembolization-Induced Cardiac Injury by Suppressing Nox2-Induced ROS Overproduction and Myocardial Apoptosis. Cardiovasc Toxicol 22, 341–351 (2022). https://doi.org/10.1007/s12012-021-09716-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-021-09716-4