
Abstract
Insulin increases glucose uptake and metabolism in skeletal muscle by signal transduction via protein phosphorylation cascades. Insulin action on signal transduction is impaired in skeletal muscle from Type 2 diabetic subjects, underscoring the contribution of molecular defects to the insulin resistant phenotype. This review summarizes recent work to identify downstream intermediates in the insulin signaling pathways governing glucose homeostasis, in an attempt to characterize the molecular mechanism accounting for skeletal muscle insulin resistance in Type 2 diabetes. Furthermore, the effects of pharmaceutical treatment of Type 2 diabetic patients on insulin signaling and glucose uptake are discussed. The identification and characterization of pathways governing insulin action on glucose metabolism will facilitate the development of strategies to improve insulin sensitivity in an effort to prevent and treat Type 2 diabetes mellitus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
During the last decades, the study of the insulin signaling pathway to glucose uptake in skeletal muscle has lead to many breakthroughs, and has provided a better understanding of the mechanisms involved in the pathogenesis of Type 2 diabetes mellitus. The discovery of novel targets within the insulin signaling cascades and the identification of protein interactions and feedback loops has underscored the complexity of intracellular signaling systems. The design of pharmaceutical approaches to prevent and treat insulin resistance will require extensive biological understanding of the signaling networks controlling glucose homeostasis. Undoubtedly, prevention and treatment efforts for the rapid global increase in obesity, Type 2 diabetes and the metabolic syndrome are a major challenge for the medical research field in the nearest future. This review is focused on describing recent work to delineate insulin signaling defects in Type 2 diabetes.
Type 2 diabetes—metabolic disorder characterized by insulin resistance
Glucose is a major fuel for skeletal muscle and other tissues. In the postprandial state after a meal, insulin is secreted from the pancreatic β-cells into the circulation to promote glucose uptake into insulin-sensitive tissues and to decrease the endogenous hepatic glucose production. When glucose levels are low, the pancreatic α-cells releases glucagon into the circulation that promotes glucose production from the liver. This system is tightly regulated to achieve glucose homeostasis (Fig. 1). Type 2 diabetes is a metabolic disorder characterized by insulin resistance along pathways governing peripheral glucose uptake and hepatic glucose production. As a result, glucose levels in the circulation increase and hyperglycemia develops. Chronic hyperglycemia is associated with many serious pathological complications and causes a variety of macrovascular and microvascular complications, such as atherosclerosis, nephropathy, neuropathy, and retinopathy. Thus, to prevent these complications euglycemia should be maintained.

Maintenance of glucose homeostasis. High glucose levels in the circulation trigger the pancreas to release insulin. Insulin release leads to an increased (+) glucose uptake in skeletal muscle and adipose tissue, and a decreased (−) glucose production from the liver. Low glucose levels lead to glucagon release to increase (+) glucose production from the liver
Skeletal muscle—role in whole body metabolism
Glucose can either undergo oxidative or non-oxidative metabolism in skeletal muscle. Glucose is oxidized by glycolytic processes generating pyruvic acid that is subsequently transformed to acetyl-CoA that is further metabolized in the citric acid cycle and that ultimately leads to generation of chemically bound energy in form of ATP. If glucose is not immediately utilized for energy, it undergoes non-oxidative metabolism and is stored as glycogen, which can be utilized for energy production later when the muscle cell requires energy.
During a euglycemic hyperinsulinemic clamp, peripheral tissues accounts for approximately 80–90% of glucose disposal, where skeletal muscle tissue is quantitatively the most important tissue [1, 2]. However, in the postprandial period after ingestion of a meal, skeletal muscle accounts for approximately 1/3 of the ingested glucose [3, 4]. Notably, skeletal muscle is one of the key tissues involved in the maintenance of whole-body glucose homeostasis, and insulin resistance in skeletal muscle is a major contributor to the pathogenesis of Type 2 diabetes.
Glucose transport—rate limiting step for glucose metabolism
Glucose is a hydrophilic molecule and thus, cannot diffuse through the lipid bilayer of cell surface membrane. Therefore, glucose entry into the cell needs to be facilitated by membrane transporters. There are mainly two different families of glucose transporters in humans, the sodium dependent glucose transporter family and the facilitative glucose transporters (GLUT) [5, 6]. While the sodium dependent glucose co-transporters are mainly involved in glucose absorption in the intestine and the kidneys, GLUT family members are facilitative glucose transporters. At present there are 14 identified genes coding for individual proteins of the GLUT family (GLUT1-14). The GLUT’s have different tissue expression and different affinity for glucose [6]. In skeletal muscle GLUT4 is the predominantly expressed isoform [7–9] and GLUT4 is localized intercellular and is translocated to the surface membrane in response to insulin, exercise, or hypoxia [10–13].
Impaired insulin-stimulated glucose transport in skeletal muscle is the rate determining step in the reduced glycogen synthesis observed in insulin resistant Type 2 diabetic patients [14]. Total GLUT4 expression is not reduced in skeletal muscle from Type 2 diabetic patients [15–17]. Thus, impaired glucose uptake in insulin resistant skeletal muscle cannot be explained by a decrease in production of GLUT4, rather impairments in insulin signaling are likely to play a role.
Early studies in adipocytes revealed that the glucose transport system was translocated from an intracellular compartment to the cell surface membrane in response to insulin [18, 19]. Later a unique insulin-regulatable glucose transport protein was identified in skeletal muscle and adipose tissue [20], which was subsequently cloned and characterized as GLUT4 [8, 9]. GLUT4 was shown to be the glucose transporter that accounts for the major part of insulin-induced glucose uptake in cultured adipocytes [21]. Using a bis-mannose photolabel technique, a strong correlation between insulin-induced 3-O-methyl-glucose uptake and cell surface expression of GLUT4 was observed [21]. GLUT4 is a membrane protein and resides within intracellular membrane bound vesicles. In response to insulin or exercise (Fig. 2) these GLUT4 containing vesicles are translocated to fuse with the cell surface membrane to facilitate transport of glucose molecules into the cell [22]. Translocation and trafficking of GLUT4 containing vesicles is impaired in insulin resistant human skeletal muscle [13, 23]. Currently whether defects in GLUT4 translocation precede the development of Type 2 diabetes is unknown.

Principle of insulin-induced glucose uptake in skeletal muscle. Insulin binds to the insulin receptor (1), thereby initiating a signaling cascade through the insulin signaling pathway (2). The intracellular membrane bound GLUT4 containing vesicles are then translocated to the cell surface membrane (3), where they ultimately integrate GLUT4 in the membrane (4) which then facilitates glucose transport into the muscle cell (5)
Insulin signaling
Insulin-responsive tissues express insulin receptors (IR) at the cell surface plasma membrane. The IR consists of four subunits, two extracellular insulin-binding α-peptides linked with two transmembrane β-peptides. On the β-subunits intracellular side, there is a tyrosine kinase domain. The β-subunits are activated due to autophosphorylation when insulin binds to the receptor. Phosphorylated tyrosine residues on the activated insulin receptor protein provide docking sites for the binding of several down-stream signaling molecules including Shc, Grb2, and the insulin receptor substrate (IRS) proteins. Insulin increases signal transduction along metabolic and mitogenic/gene-regulatory pathways (Fig. 3).

Insulin signaling along metabolic pathways. Components of the insulin signaling pathway to glucose transport are reviewed in text
Activated IR recruits IRS, which binds to phosphorylated tyrosine residues on the receptor via a phosphotyrosine binding (PTB) domain on IRS. When IRS is bound to IR, the kinase activity of IR can catalyze phosphorylation of tyrosine residues on IRS. IRS’s are scaffolding proteins and the two most predominant IRS’s involved in metabolic regulation in human skeletal muscle are IRS-1 and IRS-2. These IRS’s share a high sequence similarity, but appear to have specific signaling roles. In L6 myotubes, where IRS-1 or IRS-2 protein expression was reduced approximately 75% using small interfering RNA-mediated gene silencing, IRS-1 was demonstrated to be responsible for reduction in glucose uptake and GLUT4 translocation, whereas IRS-2 was without effect on these responses [24]. There is also direct evidence for tissue specific effects of the IRS’s from functional genomics. In transgenic mice, where either a heterozygous null mutation of genes coding IR/IRS-1 or IR/IRS-2 are expressed, the former developed severe insulin resistance in skeletal muscle, while the latter developed severe insulin resistance in liver, with only mild insulin resistance in skeletal muscle [25]. Additional evidence for tissue specific differences in IRS action is provided from studies in which IRS-2 knockout mice develop Type 2 diabetes, partly by becoming insulin resistant, but primarily because of reduced β-cell mass, which therefore prevents an adequate compensation in insulin release [26].
IRS/PI 3-kinase signaling
Tyrosine-phosphorylated IRS-1 mediates the insulin signaling to downstream enzymes by binding to a number of src-homology 2 (SH2) domain-containing signaling proteins. One important signaling intermediate that promotes the insulin signal is the phosphatidylinositol 3 (PI 3)-kinase [27]. PI 3-kinase is composed of one regulatory subunit and one catalytic subunit, which phosphorylates the 3’-OH position of the inositol ring of plasma membrane inositol phospholipids. The substrate products that are generated are phosphatidylinositol-3-monophosphate (PtdIns(3)P), PtdIns-3,4-bisphosphate (PtdIns(3,4)P2), and PtdIns-3,4,5-triphosphate (PtdIns(3,4,5)P3). These phosphoinositides are specifically recognized by proteins that contain pleckstrin homology (PH) domains, which are then redistributed to the plasma membrane.
Signaling downstream of PI 3-kinase
The serine/threonine kinase Akt, also known as protein kinase B (PKB), is a central intermediate for many of the insulin and growth factor responses downstream of PI 3-kinase. Akt was identified to be one of the insulin-responsive kinases that phosphorylates glycogen synthase kinase-3 (GSK-3) [28]. Subsequent studies demonstrated that Akt has a role in promoting GLUT4 translocation in adipocytes [29] and glucose transport and glycogen synthesis in L6 myotubes [30].
Subsequently to insulin stimulation, the generation of PtdIns(3,4,5)P3 by PI 3-kinase is necessary for re-localization of Akt to the cell surface membrane via interaction of its N-terminal PH domain. Akt was characterized to be activated by two phosphorylation steps in response to insulin [31]. Akt is phosphorylated at Ser473 in a hydrophobic motif at the C-terminal tail. DNA-dependent protein kinase (DNA-PK) [32] or the mTOR:Rictor:GβL complex [33] is responsible for Akt phosphorylation at Ser473. Thus, the true identity of this kinase or complex of protein kinases is unclear [34]. Akt is phosphorylated at Thr308 in the catalytic domain by 3′-phosphoinositide-dependent kinase 1 (PDK1) to achieve full kinase activity. PDK1 interacts with the second messengers products of PI 3-kinse, PtdIns(3,4,5)P3, and PtdIns(3,4)P2, with its PH domain and is believed to localize in the proximity of Akt [35, 36].
Three isoforms of Akt (Akt1, Akt2, and Akt3) are expressed in skeletal muscle. Experimental work with knockout mice deficient of either Akt1 or Akt2 demonstrates that there are isoform-specific roles [37, 38]. Akt2 deficient mice have an impaired ability of insulin to lower blood glucose because of defects in the action of the hormone on liver and skeletal muscle, thus Akt2 is essential in the maintenance of normal glucose homeostasis [38]. In contrast, Akt1 is required for normal growth, but dispensable for maintenance of glucose homeostasis in mice [37]. Other signaling proteins downstream of PI 3-kinase involved in insulin signaling to glucose transport include the protein kinase C-ζ and λ isoforms (PKC-ζ/λ). PKC-ζ/λ belong to the atypical family of PKC’s and are phosphorylated and activated by PDK1 [39]. The involvement of PKC-ζ in insulin signaling to glucose uptake in skeletal muscle was first investigated in L6 myotubes, where immunoprecipitable PKC-ζ activity was inhibited by the PI 3-kinase inhibitor wortmannin [40]. Furthermore, a stable and transient expression of a kinase-inactive PKC-ζ inhibited basal and insulin-stimulated glucose transport in L6 myotubes [40].
Linking insulin signaling and glucose transport
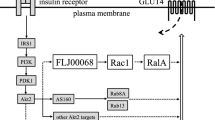
There has been great progress in elucidating the insulin signaling pathway leading to GLUT4 translocation and glucose uptake in skeletal muscle cells. In addition, the understanding of mechanisms involved in the trafficking of GLUT4 containing vesicles and their fusion with the cell surface plasma membrane has also been evolving. The post-receptor signal transduction events that are initiated when insulin binds to its receptor which are mediated primarily by kinase/phosphoryation action have been established during recent years. There is a high level of complexity within the canonical insulin signaling pathways, implying that signaling networks, rather than signaling pathways, mediate downstream responses. One of the primarily metabolic roles of insulin is to promote translocation of intracellular GLUT4 to the cell surface membrane to facilitate glucose uptake into the cell. However, the link between the insulin signaling cascade and the translocation of the GLUT4-contaning vesicles to the plasma membrane has been elusive. Different members of the small GTPase protein families have been proposed to be required for membrane trafficking of GLUT4, with members of the Rab family of GTPases under particular investigation [41].
AS160
Recently a novel Akt substrate of 160 kDa (AS160) was identified in 3T3-L1 adipocytes and implicated to play a role in glucose transport [42]. This protein contains a Rab GTPase activating protein (GAP) domain and is phosphorylated on several serine and threonine residues in response to insulin. In untreated adipocytes AS160 is concentrated in the low density microsomes (LDM) fraction. Stimulation with insulin caused a marked redistribution of AS160 from the LDM compartment to the cytosol. When several of the serine and threonine residues, which are phosphorylated in response to insulin, are mutated to alanine, GLUT4 translocation is inhibited in 3T3-L1 adipocytes [43, 44]. This inhibition did not occur when the GAP function was inactivated by a point mutation [43], indicating AS160 is required for GLUT4 vesicle translocation. mRNA of AS160, also designated as KIAA0603 (TBC1 domain family member 4), is highly expressed in human heart and skeletal muscle tissue compared to other tissues [45]. In addition, AS160 is phosphorylated in response to insulin in a dose-dependent fashion in rat skeletal muscle [46, 47]. The proposed mechanism for this action is that phosphorylation of AS160 in response to insulin leads to either inactivation of the Rab GAP function or redistribution of AS160 from the LDM compartment, which thereby activates the Rab proteins involved in GLUT4 vesicle translocation. Several Rab proteins have been suggested to be targets for the GAP activity of AS160 [48]. When a recombinant GAP domain of AS160 was tested for its activity against several Rab proteins in vitro, Rab 2A, 8A, 10, and 14 were identified with the highest GTPase activity [48]. Insulin stimulation of GLUT4 exocytosis is dependent on AS160 phosphorylation, while inhibition of endocytosis is independent of AS160 [44].
Insulin signaling defects are associated with Type 2 diabetes
Several studies have provided evidence for defects at different levels in the insulin signaling cascade in human skeletal muscle using in vivo and in vitro approaches [49]. However, whether insulin signaling defects are a cause or a consequence of skeletal muscle insulin resistance in Type 2 diabetes is unknown. The published results of insulin signaling defects in skeletal muscle are somewhat inconsistent and are mainly focused on assessments of insulin action in people with long-standing Type 2 diabetes. There are obviously many parameters that can influence the outcome from these different studies, the intention here is not to discuss these potential differences in detail, but rather to give examples of reported defects observed in insulin resistant obese and Type 2 diabetic subjects.
Protein expression of IR in skeletal muscle from Type 2 diabetic patients is unaltered [50, 51]. Insulin binding to IR has been reported to be normal [52] or impaired [53, 54]. Furthermore, tyrosine phosphorylation and/or activity of the IR has been reported to be normal [51, 53, 55–57] or impaired [50, 54, 58] in skeletal muscle from non-obese and obese Type 2 diabetic subjects, compared with non-diabetic subjects.
In isolated skeletal muscle from non-obese Type 2 diabetic patients and healthy subjects incubated with insulin (0.6–60 nmol/l), tyrosine phosphorylation of IR β-subunit is similar, whereas glucose transport is impaired at all levels of insulin studied [51]. Thus, signaling defects at the level of the insulin receptor are unlikely to fully account for the impairments in glucose transport in skeletal muscle from Type 2 diabetic patients. However, in morbidly obese insulin resistant humans phosphorylation and protein expression of IR is reduced [59]. Thus, post-receptor defects are likely to account for impairments in skeletal muscle glucose uptake in diabetes.
One of the first post-receptor events in insulin signaling is IRS-1 phosphorylation. Defects in IRS-1 function have been reported in skeletal muscle from Type 2 diabetic patients. In vivo and in vitro studies reveal that insulin-stimulated tyrosine phosphorylation of IRS-1 is impaired in non-obese and obese Type 2 diabetic patients [51, 57, 60–62]. This defect is not associated with altered protein expression of IRS-1 [51, 60, 61, 63]. A growing body of evidence indicates that increased serine/threonine phosphorylation of IRS-1 can decrease the ability of IRS-1 to be tyrosine-phosphorylated by the insulin receptor. One serine residue located near the PTB domain in IRS-1 is Ser312 (Ser307 in rat IRS-1). IRS-1 can be phosphorylated by insulin-stimulated or stress-activated kinases, such as JNK on this serine residue [64], thereby disrupting the interaction between the catalytic domain of the insulin receptor and the PTB domain of IRS-1 [65]. IRS-1 Ser636 is another residue suggested to be involved in the impaired insulin signaling in Type 2 diabetes. In cultured skeletal muscle cells from Type 2 diabetic patients, increased basal phosphorylation of IRS-1 on Ser636 has been observed, providing a putative mechanism for reduced IRS-1 tyrosine phosphorylation [62]. Impaired insulin-stimulated PI 3-kinase activity in skeletal muscle from Type 2 diabetic subjects has also been reported in vivo [57, 60, 61, 63, 66] and in vitro [51, 62, 67] in skeletal muscle from lean [51, 67] and obese Type 2 diabetic patients [57, 61, 63, 66].
Little is known about the physiological regulation of PDK-1 in skeletal muscle from Type 2 diabetic patients. In a recent study, insulin action in vivo on PDK-1 activity in vastus lateralis muscle from lean, obese insulin resistant and obese Type 2 diabetic subjects has been determined [57]. Insulin increased PDK-1 activity 2-fold in skeletal muscle from healthy subjects, with a similar response noted in obese insulin resistant and Type 2 diabetic subjects. PDK-1 protein expression was similar between lean, obese insulin resistant and obese Type 2 diabetic subjects. Thus, insulin action on PDK-1 appears to be normal in insulin resistant skeletal muscle.
Downstream targets of PDK1 include the atypical PKC (aPKC) isoforms ζ and λ and Akt. Protein expression and activity of PKC-ζ/λ have been reported to be impaired in obese Type 2 diabetic patients in vivo [57, 63]. Protein expression and activity of Akt is reported to be normal in obese Type 2 diabetic patients in vivo [63, 66]. In contrast with these in vivo experiments, in vitro studies indicate that insulin-stimulated Akt activity is reduced in skeletal muscle from Type 2 diabetic subjects stimulated with pharmacological levels of insulin [68]. However, Akt activity was normal when skeletal muscle strips were incubated with a lower concentration of insulin [68]. Since three Akt isoforms are expressed in insulin target tissues, a selective isoform-specific impairment may be masked by compensation from other isoforms. This has been illustrated in a study where insulin action on Akt isoforms in morbidly obese insulin resistant subjects was assessed [69]. In skeletal muscle from lean insulin sensitive subjects, insulin activated all three Akt isoforms, whereas only Akt1 activity was increased in skeletal muscle from obese subjects [69].
AS160 links the proximal insulin signaling steps to the translocation of GLUT4 vesicles [42]. Akt phosphorylates its substrate proteins at a RXRXXS/T peptide sequence motif. In humans, immunoblot analysis with an anti-phospho-Akt substrate antibody revealed several insulin-responsive phospho-proteins, where a protein of 160 kDa was the most prominent [70]. AS160 phosphorylation and protein expression under basal conditions is comparable between Type 2 diabetic patients and control subjects [70]. However, the increment of insulin-induced phosphorylation over basal state was markedly reduced in skeletal muscle from Type 2 diabetic patients compared to healthy controls. Therefore, the reduction in AS160 phosphorylation is not caused by reduced protein expression, but rather impaired signaling from PI 3-kinase. Our results provide evidence that impaired insulin action on AS160 is likely to account for GLUT4 translocation and glucose uptake defects in skeletal muscle from Type 2 diabetic patients.
Insulin action on Akt
Given that AS160 phosphorylation is impaired in Type 2 diabetic patients [70], phosphorylation status of the serine/threonine protein kinase Akt was determined. When site specific phosphorylation of Akt is determined, insulin-induced Ser473-phosphorylation is comparable between Type 2 diabetic patients and control subjects, whereas Thr308-phosphorylation over basal is reduced in Type 2 diabetic patients [70]. The discrepancy between observed unchanged Akt activity [66, 68], and reduced Thr308-phosphorylation [70] is unclear. Isoform-specific impairments in insulin action of Akt2 and Akt3, but not Akt1, have been observed in skeletal muscle from morbidly obese insulin resistant subjects [69]. Thus, Akt isoform-specific defects could potentially account for the differences in insulin action on Akt activity and phosphorylation, as assessed in our moderately obese diabetic cohort. Alternatively, the impairment in insulin-induced Thr308-phosphorylation may be a consequence of the altered metabolic milieu, as the Type 2 diabetic patients are often studied under hyperglycemic conditions. Consistent with this hypothesis, an earlier study revealed insulin-induced Thr308-phosphorylation was unchanged between control subjects and Type 2 diabetic patients after an overnight normalization of glycemia [56].
Link between insulin signaling and Glut4 translocation
A proposed hypothesis for the involvement of AS160 in mediating translocation of GLUT4 containing vesicles to the plasma membrane is based on evidence from experiments using different mutated forms of AS160 [43, 44]. This hypothesis is based on the fact that AS160 contains a GAP domain for Rab proteins [42], which are small G proteins involved in GLUT4 membrane trafficking [41]. Rab proteins are in an activated form when bound to GTP and inactivated when bound to GDP. Rabs have a relatively low intrinsic GTPase activity. GAP domain-containing proteins have the ability to catalyze the conversion of GTP to GDP and thereby keep Rabs in an inactive form. The active AS160 Rab GAP domain may retain the Rab proteins in an inactive form and thereby retain GLUT4 vesicles in an intracellular compartment. When AS160 is phosphorylated in response to insulin, GAP activity may either be inhibited or alternatively AS160 is relocated from the LDM compartment, resulting in release of GLUT4 vesicles that subsequently undergo translocation to the cell surface membrane.
While the exact nature of the Rab target for AS160 is unknown, Rab4 and Rab11 have been linked to GLUT4 traffic in insulin sensitive tissues. Rab4 has been shown to co-precipitate with GLUT4 containing vesicles in intracellular membrane fractions in rat skeletal muscle [71, 72] and upon insulin stimulation, the abundance of Rab4 was decreased in intracellular membrane compartments and undetected at the plasma membrane [72]. Rab11 has been suggested to be involved in the endosomal recycling, sorting, and exocytotic movement of GLUT4 in rat cardiac muscle [73]. However, in a recent report, a recombinant GAP domain of AS160 was tested for its activity against several Rab proteins in adipocytes [48]. The study provided evidence that Rab 2A, 8A, 10, and 14 have the highest GTPase activity in response to exposure of the recombinant GAP domain of AS160. Thus, AS160 may constitute a point of convergence between insulin signaling and GLUT4 vesicular trafficking. However, the exact Rabs targeted by AS160 in human skeletal muscle GLUT4 trafficking are still unknown and clearly more work in this area is required.
Insulin signaling after anti-diabetic treatment
In clinical use today, there are several treatment approaches which are administered in order to reduce hyperglycemia in Type 2 diabetic patients. The oral anti-diabetic agent metformin (dimethyl biguanide) has the longest history in diabetes treatment. Metformin was introduced in 1957, where it was reported to have anti-diabetic properties [74]. Metformin is the most widely prescribed agent for diabetes treatment today. Despite being in clinical use for almost half a century, the identity of its cellular target is still elusive. Metformin primarily acts on the liver to inhibit glucose production [75] possibly by action via AMP-activated protein kinase (AMPK) [76].
A more recent pharmacological agent for Type 2 diabetes treatment is rosiglitazone, a drug that belongs to the Thiazolidinedione (TZD) family. TZD’s act as agonistic ligands of the nuclear hormone receptor transcription factor PPARγ. PPARγ is predominantly expressed in adipocytes, but it is also expressed in skeletal muscle [77]. Many studies have reported positive effects such as improved glycemic control after rosiglitazone treatment [78–85]. Even though the insulin sensitization effects of TZD’s are well-documented, the molecular mechanisms responsible for these effects in humans are still elusive. Thus, investigations of potential effects on insulin signaling responses and gene expression in human skeletal muscle after treatment with either metformin or rosiglitazone are required. Early treatment of Type 2 diabetic patients in an early phase of the disease progression, before the metabolic derangements become too severe, is of particular importance to facilitate attempts to hinder many of the end-stage complications related to the disease. Moreover, investigations of the treatment effects in moderately obese newly diagnosed Type 2 diabetic patients may have the potential to reveal outcomes that might be masked in obese patients with a longer duration of the disease, where many other factors would have to be taken into account caused by the more severe metabolic derangements.
Insulin signaling has been assessed in vivo in skeletal muscle before and after treatment with either metformin or rosiglitazone [86]. Newly diagnosed Type 2 diabetic patients were recruited and randomized into three groups, one treated with metformin, one with rosiglitazone, and one placebo control group. The treatment period lasted 26 weeks. Before and after the treatment period, patients were subjected to a euglycemic hyperinsulinemic clamp whereby whole-body, as well as skeletal muscle specific glucose uptake was determined. Clinical and metabolic characteristics of the study participants were assessed before and after treatment. BMI, HbA1c, and fasting plasma glucose were decreased after metformin treatment, but not after rosiglitazone or placebo treatment. Fasting FFA levels were similar between the groups and unchanged after treatment. However, post-treatment serum FFA levels measured during the hyperinsulinemic clamp were decreased after metformin and rosiglitazone treatment, respectively, and unchanged in the placebo group. Whole-body and skeletal muscle specific glucose uptake was also significantly increased after rosiglitazone treatment. In contrast, after treatment with metformin or placebo, insulin-mediated glucose uptake was unaltered. Whole-body glucose uptake was tightly correlated with glucose uptake in skeletal muscle measured with PET scan analysis over a portion of the vastus lateralis muscle, consistent with earlier work [87].
The increase in insulin sensitivity in skeletal muscle after TZD treatment may be due to an augmentation in intracellular insulin signaling events [86]. Signaling events were determined at the early post-receptor level, namely tyrosine-phosphorylation of IRS-1 and IRS-1-associated PI 3-kinase activity, since these intermediate steps have been reported to be impaired in Type 2 diabetic patients [51, 60]. Prior to treatment, insulin infusion increased IRS-1 tyrosine phosphorylation and IRS-1 associated PI 3-kinase activity. Neither metformin nor rosiglitazone treatment enhanced insulin-stimulated IRS-1 tyrosine phosphorylation nor IRS-1-associated PI 3-kinase activity. Downstream signaling events related to Akt, including Ser473-phosphoryaltion of Akt and phosphorylation of the Akt substrate AS160 were also assessed. Before the treatment period, insulin stimulation increased Ser473-phosphorylation and AS160 2 to 3-fold, with similar effects between the rested and exercised leg. However, neither metformin nor rosiglitazone treatment enhanced phosphorylation of Akt nor AS160 [86].
Metformin treatment improved HbA1c and fasting blood glucose levels, but was without effect on insulin-mediated glucose uptake [86]. This is consistent with previous studies of poorly controlled obese Type 2 diabetic patients [88, 89]. Previous studies have provided evidence that improvements in glucose homeostasis after metformin treatment are primarily due to an insulin-sensitizing effect on the liver, leading to a decrease in hepatic glucose production [88–90]. Furthermore, increased rates of insulin-stimulated whole-body glucose disposal have been reported [90, 91]. However, the increase in glucose disposal rate was minor and may potentially be explained by an improvement in glucose homeostasis.
In contrast to metformin treatment in the present study, rosiglitazone treatment enhanced insulin-mediated whole body and leg muscle glucose uptake, but was without effect on HbA1c and fasting blood glucose. The improvement in insulin-mediated glucose uptake reported here is consistent with previous studies whereby either troglitazone [91, 92] or rosiglitazone [63, 93, 94] was administered, further supporting the insulin sensitizing effect on skeletal muscle. However the lack of an improvement in HbA1c and fasting blood glucose is an inconsistent finding, since both improved [91, 93–95] and unchanged [78, 96] glycemic control has been observed in Type 2 diabetic patients undergoing TZD treatment.
The insulin-sensitizing effect of rosiglitazone on glucose uptake in skeletal muscle is unrelated to changes in insulin signaling at the intermediate steps. This is an unexpected finding since similar studies have been performed whereby positive effects of TZD-treatment on insulin signaling intermediates have been reported. For example, a previous study provides evidence that treatment of poorly controlled obese Type 2 diabetic patients with metformin for 3–4 months enhanced insulin-mediated glucose uptake independent of improved insulin signaling [91]. However, in contrast to our results [86], treatment with troglitazone for 3–4 months improved insulin-mediated whole body glucose uptake concomitant with improved insulin action on PI 3-kinase and Akt [91]. Furthermore, treatment of obese Type 2 diabetic subjects for 1 month with rosiglitazone also enhanced insulin-mediated whole body glucose uptake, coincident with increased IRS-1 associated PI 3-kinase and atypical PKC activity [63]. These studies imply the insulin-sensitizing effect of TZD treatment in obese Type 2 diabetic subjects is partly caused by enhanced peripheral insulin action.
Conclusions and future perspectives
The molecular mechanisms of insulin signal transduction to glucose transport in insulin resistant human skeletal muscle have been addressed in this review. We summarize new data on AS160, the most proximal step identified in the insulin signaling transduction pathway leading to GLUT4 translocation. Experimental evidence from studies in adipocytes reveals that AS160 is required for insulin-induced translocation of GLUT4 to the cell surface plasma membrane in order to facilitate glucose uptake. Given the role of AS160 in insulin-mediated GLUT4 translocation in adipocytes, our data suggest that aberrant insulin signaling to IRS-1/PI 3-kinase/Akt/AS160 contributes to defects in GLUT4 translocation and glucose uptake in skeletal muscle from insulin resistant Type 2 diabetic patients. Impaired insulin action on AS160 is associated with reduced Akt Thr308-phosphorylation, suggesting that these two events are linked. Since the protein expression of AS160 was unaltered in Type 2 diabetic patients, the reduced AS160 phosphorylation is likely a result from impaired upstream signaling, increased Ser/Thr-phosphatase activity, or both.
In newly diagnosed Type 2 diabetic patients treated with rosiglitazone skeletal muscle glucose uptake is improved, independent of improved insulin signaling at the level of IRS-1/PI 3-kinase/Akt/AS160. This finding raises the possibility that even more distal events in the GLUT4 translocation machinery are augmented after rosiglitazone treatment (Fig. 4). Further studies are necessary to investigate GLUT4 vesicular trafficking and vesicular fusion machinery in skeletal muscle of Type 2 diabetic patients after rosiglitazone treatment. The identification of the molecular mechanism of action by which TZD treatment enhances insulin sensitivity in tissues is important from at least two perspectives; first, resolution of the cellular mechanism of action will eventually facilitate more specific design of drug compounds, and second, the identification of the mechanism of action of TZD treatment will also potentially give important clues to unravel the pathophysiology of Type 2 diabetes.

Rosiglitazone treatment increased glucose uptake in skeletal muscle independent of enhancement in insulin signaling pathway. Distal components at the level of GLUT4 translocation and/or fusion with the cell surface membrane are likely to be augmented
To achieve the most efficient and manageable treatment of Type 2 diabetes, an improved understanding of the underlying defects is crucial. Identification of defects in the insulin-glucose-homeostatic relationship requires deeper knowledge of the molecular mechanisms which elicit specific physiological events. This review highlights the importance of designing treatment strategies for insulin resistance in skeletal muscle to target at multiple sites depending on the progression of the disease. In overt Type 2 diabetes, where insulin resistance is attenuated by the derangements in the metabolic milieu, insulin sensitivity in skeletal muscle is important to regain by augmentation of defects in the insulin signaling pathway. In newly diagnosed Type 2 diabetic patients with relatively good metabolic control, the improvement of glucose uptake by TZD treatment is achieved through improvements distal in the signaling pathway, at the level of GLUT4 translocation. Therefore, more intensified research of the distal mechanism of GLUT4 translocation and trafficking is desirable in the future.
References
DeFronzo, R. A., Jacot, E., Jequier, E., Maeder, E., Wahren, J., & Felber, J. P. (1981). The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes, 30, 1000–1007.
DeFronzo, R. A., Gunnarsson, R., Bjorkman, O., Olsson, M., & Wahren, J. (1985). Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (Type II) diabetes mellitus. Journal of Clinical Investigation, 76, 149–155.
Kelley, D., Mitrakou, A., Marsh, H., Schwenk, F., Benn, J., Sonnenberg, G., Arcangeli, M., Aoki, T., Sorensen, J., Berger, M., Sonksen, P., & Gerich, J. (1988). Skeletal muscle glycolysis, oxidation, and storage of an oral glucose load. Journal of Clinical Investigation, 81, 1563–1571.
Moore, M. C., Cherrington, A. D., & Wasserman, D. H. (2003). Regulation of hepatic and peripheral glucose disposal. Best Practice & Research. Clinical Endocrinology & Metabolism, 17, 343–364.
Joost, H.-G., Bell, G. I., Best, J. D., Birnbaum, M. J., Charron, M. J., Chen, Y. T., Doege, H., James, D. E., Lodish, H. F., Moley, K. H., Moley, J. F., Mueckler, M., Rogers, S., Schurmann, A., Seino, S., & Thorens, B. (2002). Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. American Journal of Physiology. Endocrinology and Metabolism, 282, E974–E976.
Scheepers, A., Joost, H. G., & Schurmann, A. (2004). The glucose transporter families SGLT and GLUT: Molecular basis of normal and aberrant function. Journal of Parenteral and Enteral Nutrition, 28, 364–371.
Birnbaum, M. J. (1989). Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell, 57, 305–315.
Fukumoto, H., Kayano, T., Buse, J. B., Edwards, Y., Pilch, P. F., Bell, G. I., & Seino, S. (1989). Cloning and characterization of the major insulin-responsive glucose transporter expressed in human skeletal muscle and other insulin-responsive tissues. Journal of Biological Chemistry, 264, 7776–7779.
James, D. E., Strube, M., & Muecdler, M. (1989). Molecular cloning and characterization of an insulin-regulatable glucose transporter. Nature, 338, 83–87.
Douen, A. G., Ramlal, T., Rastogi, S., Bilan, P. J., Cartee, G. D., Vranic, M., Holloszy, J. O., & Klip, A. (1990). Exercise induces recruitment of the “insulin-responsive glucose transporter”. Evidence for distinct intracellular insulin- and exercise- recruitable transporter pools in skeletal muscle. Journal of Biological Chemistry, 265, 13427–13430.
Hirshman, M. F., Goodyear, L. J., Wardzala, L. J., Horton, E. D., & Horton, E. S. (1990). Identification of an intracellular pool of glucose transporters from basal and insulin-stimulated rat skeletal muscle. Journal of Biological Chemistry, 265, 987–991.
Kristiansen, S., Hargreaves, M., & Richter, E. A. (1996). Exercise-induced increase in glucose transport, G. L.,UT-4, and VAMP-2 in plasma membrane from human muscle. American Journal of Physiology. Endocrinology and Metabolism, 270, E197-201.
Ryder, J., Yang, J., Galuska, D., Rincon, J., Bjornholm, M., Krook, A., Lund, S., Pedersen, O., Wallberg-Henriksson, H., Zierath, J. R., & Holman, G (2000). Use of a novel impermeable biotinylated photolabeling reagent to assess insulin- and hypoxia-stimulated cell surface GLUT4 content in skeletal muscle from type 2 diabetic patients. Diabetes, 49, 647–654.
Cline, G. W., Petersen, K. F., Krssak, M., Shen, J., Hundal, R. S., Trajanoski, Z., Inzucchi, S., Dresner, A., Rothman, D. L., & Shulman, G. I. (1999). Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in Type 2 diabetes. The New England Journal of Medicine, 341, 240–246.
Handberg, A., Vaag, A., Damsbo, P., Beck-Nielsen, H., & Vinten, J (1990). Expression of insulin regulatable glucose transporters in skeletal muscle from type 2 (non-insulin-dependent) diabetic patients. Diabetologia, 33, 625–627.
Pedersen, O., Bak, J. F., Andersen, P. H., Lund, S., Moller, D. E., Flier, J. S., & Kahn, B. B. (1990). Evidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDM. Diabetes, 39, 865–870.
Shepherd, P. R., & Kahn, B. B. (1999). Glucose transporters and insulin action: Implications for insulin resistance and diabetes mellitus. The New England Journal of Medicine, 341, 248–257.
Cushman, S. W., & Wardzala, L. J. (1980). Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. Journal of Biological Chemistry, 255, 4758–4762.
Suzuki, K., & Kono, T (1980). Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proceedings of the National Academy of Sciences of the United States of America, 77, 2542–2545.
James, D. E., Brown, R., Navarro, J., & Pilch, P. F. (1988). Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature, 333, 183–185.
Holman, G. D., Kozka, I. J., Clark, A. E., Flower, C. J., Saltis, J., Habberfield, A. D., Simpson, I. A., & Cushman, S. W. (1990). Cell surface labeling of glucose transporter isoform GLUT4 by bis- mannose photolabel. Correlation with stimulation of glucose transport in rat adipose cells by insulin and phorbol ester. Journal of Biological Chemistry, 265, 18172–18179.
Holman, G. D., & Sandoval, I. V. (2001). Moving the insulin-regulated glucose transporter GLUT4 into and out of storage. Trends in Cell Biology, 11, 173–179.
Garvey, W. T., Maianu, L., Zhu, J.-H., Brechtel-Hook, G., Wallace, P., & Baron, A. D. (1998). Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. Journal of Clinical Investigation, 101, 2377–2386.
Huang, C., Thirone, A. C. P., Huang, X., & Klip, A (2005). Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in L6 myotubes. Journal of Biological Chemistry, 280, 19426–19435.
Kido, Y., Burks, D. J., Withers, D., Bruning, J. C., Kahn, C. R., White, M. F., & Accili, D (2000). Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. Journal of Clinical Investigation, 105, 199–205.
Withers, D. J., Gutierrez, J. S., Towery, H., Burks, D. J., Ren, J.-M., Previs, S., Zhang, Y., Bernal, D., Pons, S., Shulman, G. I., Bonner-Weir, S., & White, M. F. (1998). Disruption of IRS-2 causes type 2 diabetes in mice. Nature, 391, 900–904.
Cantrell, D. A. (2001). Phosphoinositide 3-kinase signalling pathways. Journal of Cell Science, 114, 1439–1445.
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., & Hemmings, B. A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature, 378, 785–789.
Tanti, J. F., Grillo, S., Gremeaux, T., Coffer, P. J., Van Obberghen, E., & Le Marchand-Brustel, Y (1997). Potential role of protein kinase B in glucose transporter 4 translocation in adipocytes. Endocrinology, 138, 2005–2010.
Ueki, K., Yamamoto-Honda, R., Kaburagi, Y., Yamauchi, T., Tobe, K., Burgering, B. M. T., Coffer, P. J., Komuro, I., Akanuma, Y., Yazaki, Y., & Kadowaki, T (1998). Potential role of protein kinase B in insulin-induced glucose transport, glycogen synthesis, and protein synthesis. Journal of Biological Chemistry, 273, 5315–5322.
Alessi, D. R., Andjelkovic, M., Caudwell, B., Cron, P., Morrice, N., Cohen, P., & Hemmings, B. A. (1996). Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO Journal, 15, 6541–6551.
Feng, J., Park, J., Cron, P., Hess, D., & Hemmings, B. A. (2004). Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent Protein Kinase. Journal of Biological Chemistry, 279, 41189–41196.
Sarbassov, D. D., Guertin, D. A., Ali, S. M., & Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science, 307, 1098–1101.
Dong, L. Q., & Liu, F. (2005). PDK2: The missing piece in the receptor tyrosine kinase signaling pathway puzzle. American Journal of Physiology. Endocrinology and Metabolism, 289, E187–196.
Alessi, D. R., James, S. R., Downes, C. P., Holmes, A. B., Gaffney, P. R., Reese, C. B., & Cohen, P (1997). Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Current Biology, 7, 261–269.
Mora, A., Komander, D., van Aalten, D. M. F., & Alessi, D. R. (2004). PDK1, the master regulator of AGC kinase signal transduction. Seminars in Cell & Developmental Biology, 15, 161–170.
Cho, H., Thorvaldsen, J. L., Chu, Q., Feng, F., & Birnbaum, M. J. (2001). Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. Journal of Biological Chemistry, 276, 38349–38352.
Cho, H., Mu, J., Kim, J. K., Thorvaldsen, J. L., Chu, Q., Crenshaw, E. B. III, Kaestner, K. H., Bartolomei, M. S., Shulman, G. I., & Birnbaum, M. J. (2001). Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBbeta). Science, 292, 1728–1731.
Chou, M. M., Hou, W., Johnson, J., Graham, L. K., Lee, M. H., Chen, C. S., Newton, A. C., Schaffhausen, B. S., & Toker, A (1998). Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Current Biology, 8, 1069–1077.
Bandyopadhyay, G., Standaert, M. L., Galloway, L., Moscat, J., & Farese, R. V. (1997). Evidence for involvement of protein kinase C (PKC)-zeta and noninvolvement of diacylglycerol-sensitive PKCs in insulin-stimulated glucose transport in L6 myotubes. Endocrinology, 138, 4721–4731.
Cormont, M., & Le Marchand-Brustel, Y (2001). The role of small G-proteins in the regulation of glucose transport. Molecular Membrane Biology, 18, 213–220.
Kane, S., Sano, H., Liu, S. C. H., Asara, J. M., Lane, W. S., Garner, C. C., & Lienhard, G. E. (2002). A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-Activating Protein (GAP) domain. Journal of Biological Chemistry, 277, 22115–22118.
Sano, H., Kane, S., Sano, E., Miinea, C. P., Asara, J. M., Lane, W. S., Garner, C. W., & Lienhard, G. E. (2003). Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. Journal of Biological Chemistry, 278, 14599–14602.
Zeigerer, A., McBrayer, M. K., & McGraw, T. E. (2004). Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Molecular Biology of the Cell, 15, 4406–4415.
Matsumoto, Y., Imai, Y., Lu Yoshida, N., Sugita, Y., Tanaka, T., Tsujimoto, G., Saito, H., & Oshida, T (2004). Upregulation of the transcript level of GTPase activating protein KIAA0603 in T cells from patients with atopic dermatitis. FEBS Letters, 572, 135–140.
Arias, E. B., Kim, J., & Cartee, G. D. (2004). Prolonged incubation in PUGNAc results in increased protein O-linked glycosylation and insulin resistance in rat skeletal muscle. Diabetes, 53, 921–930.
Bruss, M. D., Arias, E. B., Lienhard, G. E., & Cartee, G. D. (2005). Increased phosphorylation of Akt Substrate of 160 kDa (AS160) in rat skeletal muscle in response to insulin or contractile activity. Diabetes, 54, 41–50.
Miinea, C. P., Sano, H., Kane, S., Sano, E., Fukuda, M., Peranen, J., Lane, W. S., & Lienhard, G. E. (2005). AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase activating protein domain. Biochemistry Journal, 391, 87–93.
Leng, Y., Karlsson, H. K., & Zierath, J. R. (2004). Insulin signaling defects in type 2 diabetes. Reviews in Endocrine & Metabolic Disorders, 5, 111–117.
Arner, P., Pollare, T., Lithell, H., & Livingston, J. N. (1987). Defective insulin receptor tyrosine kinase in human skeletal muscle in obesity and type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia, 30, 437–440.
Krook, A., Bjornholm, M., Galuska, D., Jiang, X., Fahlman, R., Myers, M., Wallberg-Henriksson, H., & Zierath, J. R. (2000). Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes, 49, 284–292.
Ciaraldi, T., Carter, L., Rehman, N., Mohideen, P., Mudaliar, S., & Henry, R (2002). Insulin and insulin-like growth factor-1 action on human skeletal muscle: Preferential effects of insulin-like growth factor-1 in type 2 diabetic subjects. Metabolism, 51, 1171–1179.
Caro, J. F., Sinha, M. K., Raju, S. M., Ittoop, O., Pories, W. J., Flickinger, E. G., Meelheim, D., & Dohm, G. L. (1987). Insulin receptor kinase in human skeletal muscle from obese subjects with and without noninsulin dependent diabetes. Journal of Clinical Investigation, 79, 1330–1337.
Maegawa, H., Shigeta, Y., Egawa, K., & Kobayashi, M (1991). Impaired autophosphorylation of insulin receptors from abdominal skeletal muscles in non-obese subjects with NIDDM. Diabetes, 40, 815–819.
Klein, H. H., Vestergaard, H., Kotzke, G., & Pedersen, O (1995). Elevation of serum insulin concentration during euglycemic hyperinsulinemic clamp studies leads to similar activation of insulin receptor kinase in skeletal muscle of subjects with and without NIDDM. Diabetes, 44, 1310–1317.
Meyer, M. M., Levin, K., Grimmsmann, T., Beck-Nielsen, H., & Klein, H. H. (2002). Insulin signalling in skeletal muscle of subjects with or without Type II-diabetes and first degree relatives of patients with the disease. Diabetologia, 45, 813–822.
Kim, Y.-B., Kotani, K., Ciaraldi, T. P., Henry, R. R., & Kahn, B. B. (2003). Insulin-stimulated protein kinase C lambda/zeta activity is reduced in skeletal muscle of humans with obesity and Type 2 diabetes: Reversal with weight reduction. Diabetes, 52, 1935–1942.
Nolan, J. J., Freidenberg, G., Henry, R., Reichart, D., & Olefsky, J. M. (1994). Role of human skeletal muscle insulin receptor kinase in the In vivo insulin resistance of non-insulin-dependent diabetes mellitus and obesity. The Journal of Clinical Endocrinology and Metabolism, 78, 471–477.
Goodyear, L. J., Giorgino, F., Sherman, L. A., Carey, J., Smith, R. J., & Dohm, G. L. (1995). Insulin receptor phosphorylation, insulin receptor substrate-1 phosphorylation, and phosphatidylinositol 3-kinase activity are decreased in intact skeletal muscle strips from obese subjects. Journal of Clinical Investigation, 95, 2195–2204.
Björnholm, M., Kawano, Y., Lehtihet, M., & Zierath, J. R. (1997). Insulin receptor substrate-1 phosphorylation and phosphatidylinositol 3-kinase activity in skeletal muscle from NIDDM subjects after In vivo insulin stimulation. Diabetes, 46, 524–527.
Cusi, K., Maezono, K., Osman, A., Pendergrass, M., Patti, M. E., Pratipanawatr, T., DeFronzo, R. A., Kahn, C. R., & Mandarino, L. J. (2000). Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. Journal of Clinical Investigation, 105, 311–320.
Bouzakri, K., Roques, M., Gual, P., Espinosa, S., Guebre-Egziabher, F., Riou, J.-P., Laville, M., Le Marchand-Brustel, Y., Tanti, J-F., & Vidal, H (2003). Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with Type 2 diabetes. Diabetes, 52, 1319–1325.
Beeson, M., Sajan, M. P., Dizon, M., Grebenev, D., Gomez-Daspet, J., Miura, A., Kanoh, Y., Powe, J., Bandyopadhyay, G., Standaert, M. L., & Farese, R. V. (2003). Activation of protein kinase C-zeta by insulin and phosphatidylinositol-3,4,5-(PO4)3 is defective in muscle in Type 2 diabetes and impaired glucose tolerance: Amelioration by rosiglitazone and exercise. Diabetes, 52, 1926–1934.
Lee, Y. H., Giraud, J., Davis, R. J., & White, M. F. (2003). c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. Journal of Biological Chemistry, 278, 2896–2902.
Aguirre, V., Werner, E. D., Giraud, J., Lee, Y. H., Shoelson, S. E., & White, M. F. (2002). Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. Journal of Biological Chemistry, 277, 1531–1537.
Kim, Y.-B., Nikoulina, S. E., Ciaraldi, T. P., Henry, R. R., & Kahn, B. B. (1999). Normal insulin-dependent activation of Akt/protein kinase B., with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. Journal of Clinical Investigation, 104, 733–741.
Tsuchida, H., Björnholm, M., Fernström, M., Galuska, D., Johansson, P., Wallberg-Henriksson, H., Zierath, J., Lake, S., & Krook, A (2002). Gene expression of the p85a regulatory subunit of phosphatidylinositol 3-kinase in skeletal muscle from type 2 diabetic subjects. Pflugers Archive European Journal of Physiology, 445, 25–31.
Krook, A., Roth, R., Jiang, X., Zierath, J. R., & Wallberg-Henriksson, H (1998). Insulin-stimulated Akt kinase activity is reduced in skeletal muscle from NIDDM subjects. Diabetes, 47, 1281–1286.
Brozinick, J. T. Jr, Roberts, B. R., & Dohm, G. L. (2003). Defective signaling through Akt-2 and -3 but not Akt-1 in insulin-resistant human skeletal muscle: Potential role in insulin resistance. Diabetes, 52, 935–941.
Karlsson, H. K. R., Zierath, J. R., Kane, S., Krook, A., Lienhard, G. E., & Wallberg-Henriksson, H (2005). Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of Type 2 diabetic subjects. Diabetes, 54, 1692–1697.
Aledo J. C., Darakhshan F., & Hundal H. S. (1995). Rab4, but not the transferrin receptor, is colocalized with GLUT4 in an insulin-sensitive intracellular compartment in rat skeletal muscle. Biochemical and Biophysical Research Communications, 215, 321–328.
Sherman, L., Hirshman, M., Cormont, M., Le Marchand-Brustel, Y., & Goodyear, L (1996). Differential effects of insulin and exercise on Rab4 distribution in rat skeletal muscle. Endocrinology, 137, 266–273.
Kessler, A., Tomas, E., Immler, D., Meyer, H. E., Zorzano, A., & Eckel, J (2000). Rab11 is associated with GLUT4-containing vesicles and redistributes in response to insulin. Diabetologia, 43, 1518–1527.
Bailey, C. J., & Day, C. (2004). Metformin: Its botanical background. Practial Diabetes International, 21, 115–117.
Bailey, C. J., & Turner, R. C. (1996). Metformin. The New England Journal of Medicine, 334, 574–579.
Zhou, G., Myers, R., Li, Y., Chen, Y., Shen, X., Fenyk-Melody, J., Wu, M., Ventre, J., Doebber, T., Fujii, N., Musi, N., Hirshman, M. F., Goodyear, L. J., & Moller, D. E. (2001). Role of AMP-activated protein kinase in mechanism of metformin action. Journal of Clinical Investigation, 108, 1167–1174.
Zierath, J. R., Ryder, J. W., Doebber, T., Woods, J., Wu, M., Ventre, J., Li, Z., McCrary, C., Berger, J., Zhang, B., & Moller, D. E. (1998). Role of skeletal muscle in thiazolidinedione insulin sensitizer (PPARgamma agonist) action. Endocrinology, 139, 5034–5041.
Patel, J., Anderson, R. J., & Rappaport, E. B. (1999). Rosiglitazone monotherapy improves glycaemic control in patients with type 2 diabetes: A twelve-week, randomized, placebo-controlled study. Diabetes, Obesity & Metabolism, 1, 165–172.
Fonseca, V., Rosenstock, J., Patwardhan, R., & Salzman, A (2000). Effect of metformin and rosiglitazone combination therapy in patients with type 2 diabetes mellitus: A randomized controlled trial. JAMA, 283, 1695–1702.
Nolan, J. J., Jones, N. P., Patwardhan, R., & Deacon, L. F. (2000). Rosiglitazone taken once daily provides effective glycaemic control in patients with Type 2 diabetes mellitus. Diabetic Medicine, 17, 287–294.
Raskin, P., Rappaport, E. B., Cole, S. T., Yan, Y., Patwardhan, R., & Freed, M. I. (2000). Rosiglitazone short-term monotherapy lowers fasting and post-prandial glucose in patients with type II diabetes. Diabetologia, 43, 278–284.
Wolffenbuttel, B. H., Gomis, R., Squatrito, S., Jones, N. P., & Patwardhan, R. N. (2000). Addition of low-dose rosiglitazone to sulphonylurea therapy improves glycaemic control in Type 2 diabetic patients. Diabetic Medicine, 17, 40–47.
Lebovitz, H. E., Dole, J. F., Patwardhan, R., Rappaport, E. B., & Freed, M. I. (2001). Rosiglitazone monotherapy is effective in patients with type 2 diabetes. The Journal of Clinical Endocrinology and Metabolism, 86, 280–288.
Phillips, L. S., Grunberger, G., Miller, E., Patwardhan, R., Rappaport, E. B., & Salzman, A (2001). Once- and twice-daily dosing with Rosiglitazone improves glycemic control in patients with type 2 diabetes. Diabetes Care, 24, 308–315.
Raskin, P., Rendell, M., Riddle, M. C., Dole, J. F., Freed, M. I., & Rosenstock, J (2001). A randomized trial of rosiglitazone therapy in patients with inadequately controlled insulin-treated Type 2 diabetes. Diabetes Care, 24, 1226–1232.
Karlsson, H. K., Hallsten, K., Bjornholm, M., Tsuchida, H., Chibalin, A. V., Virtanen, K. A., Heinonen, O. J., Lonnqvist, F., Nuutila, P., & Zierath, J. R. (2005). Effects of metformin and rosiglitazone treatment on insulin signaling and glucose uptake in patients with newly diagnosed type 2 diabetes: A randomized controlled study. Diabetes, 54, 1459–1467.
Zierath, J. R. (1995). In vitro studies of human skeletal muscle: Hormonal and metabolic regulation of glucose transport. Acta physiologica Scandinavica. Supplementum, 155, 1–96.
Stumvoll, M., Nurjhan, N., Perriello, G., Dailey, G., & Gerich, J. E. (1995). Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. The New England Journal of Medicine, 333, 550–554.
Hundal, R., Krssak, M., Dufour, S., Laurent, D., Lebon, V., Chandramouli, V., Inzucchi, S., Schumann, W., Petersen, K., Landau, B., & Shulman, G (2000). Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes, 49, 2063–2069.
Inzucchi, S. E., Maggs, D. G., Spollett, G. R., Page, S. L., Rife, F. S., Walton, V., & Shulman, G. I. (1998). Efficacy and metabolic effects of Metformin and Troglitazone in type II diabetes mellitus. The New England Journal of Medicine, 338, 867–873.
Kim, Y.-B., Ciaraldi, T. P., Kong, A., Kim, D., Chu, N., Mohideen, P., Mudaliar, S., Henry, R. R., & Kahn, B. B. (2002). Troglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases p110beta protein levels in skeletal muscle of type 2 diabetic subjects. Diabetes, 51, 443–448.
Petersen, K., Krssak, M., Inzucchi, S., Cline, G., Dufour, S., & Shulman, G (2000). Mechanism of troglitazone action in type 2 diabetes. Diabetes, 49, 827–831.
Carey, D. G., Cowin, G. J., Galloway, G. J., Jones, N. P., Richards, J. C., Biswas, N., & Doddrell, D. M. (2002). Effect of rosiglitazone on insulin sensitivity and body composition in Type 2 diabetic patients. Obesity Research, 10, 1008–1015.
Miyazaki, Y., He, H., Mandarino, L. J., & DeFronzo, R. A. (2003). Rosiglitazone improves downstream insulin receptor signaling in Type 2 diabetic patients. Diabetes, 52, 1943–1950.
Miyazaki, Y., Glass, L., Triplitt, C., Matsuda, M., Cusi, K., Mahankali, A., Mahankali, S., Mandarino, L. J., & DeFronzo, R. A. (2001). Effect of rosiglitazone on glucose and non-esterified fatty acid metabolism in Type II diabetic patients. Diabetologia, 44, 2210–2219.
Mayerson, A. B., Hundal, R. S., Dufour, S., Lebon, V., Befroy, D., Cline, G. W., Enocksson, S., Inzucchi, S. E., Shulman, G. I., & Petersen, K. F. (2002). The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with Type 2 diabetes. Diabetes, 51, 797–802.
Acknowledgments
The authors are supported by grants from the Swedish Medical Research Council, the Swedish Diabetes Association, Foundation for Scientific Studies of Diabetology, the Strategic Research Foundation (INGVAR), Novo-Nordisk Foundation, and the Commission of the European Communities (Network of Excellence EUGENE2; Contract No. LSHM-CT-2004-005272 and Integrated Project EXGENESIS; Contract No. LSHM-CT-2004-005272).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Karlsson, H.K.R., Zierath, J.R. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochem Biophys 48, 103–113 (2007). https://doi.org/10.1007/s12013-007-0030-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-007-0030-9