
Abstract
This study aims to clarify the molecular mechanism of fluorine exposure that leads to nerve injury. PC12 cells were treated with fluorine at different concentrations (0.5, 1.0, 1.5, and 2.0 mM). Cytoactivity was detected at different time points (2, 4, 6, 8, 12, 24, and 48 h). After 2 h, DCF was used to detect and mark the level of reactive oxygen species (ROS) within cells. After 24 h, cellular metamorphosis was observed using an inverted microscope. After 2 h, Hoechst-33342 was used to detect apoptosis. After 24 h, Western blot analysis was performed to detect apoptosis-related poly (ADP-ribose) polymerase (PARP) protein, p-elF, and expression of the endoplasmic reticulum stress-related X-box binding protein 1 (XBP-1). The results showed that Fluorine exposure resulted in a reduction of cell viability, which was negatively correlated with fluorine dose. Within certain fluorine exposure duration, the ROS level within the cell and the apoptotic level are linearly related to fluorine exposure level. XBP-1 and PARP protein are sensitive to variations in fluorine concentration, which indicates that oxidative stress from fluorine exposure can lead to apoptosis. XBP-1 and PARP may be the key proteins during the entire process. These results provide a valid basis for fluorine-induced free radical injury theory.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Long-term exposure to a high-fluorine environment may result in endemic fluorosis among susceptible populations. However, the pathogenesis of this condition remains unclear. Fluorine-induced free radical injury theory has been recognized by a large number of scholars [1]. The most common indicators used in research are SOD and MDA, which may be influenced by various factors. It does not present low specificity and sensitivity, and the obtained results contradict one another [2, 3]. On the basis of an early-stage laboratory in vivo study [4–8] and PC12 cell culture in vitro [9], an acute exposure experiment of PC12 cultivation in vitro was conducted to detect direct intracellular changes in reactive oxygen species (ROS) concentration induced by fluorine exposure at a concentration different from that in the previous study. By observing the influences of fluorine exposure on PC12 cellular morphology, viability, endoplasmic reticulum stress (ERS), and apoptosis-related proteins, the present study elucidates the molecular mechanism of ERS in neuron injury induced by fluorine exposure. The current study also determines the relation between chronergy and the quantitative efficiency of fluorine-induced neuron injury.
Materials and Methodology
Reagents
Dulbecco’s modified Eagle’s medium (a high-glucose medium), horse serum(GIBCO), defined fetal bovine serum (Hangzhou Sijiqing), trypsin, polylysine, l-glutamine, penicillin–streptomycin, Cell Counting Kit-8 (CCK-8), Hoechst-33342, ROS assay kit (Beyotime Biotechnology), bicinchoninic acid protein content detection kit, radioimmunoprecipitation assay lysate, phenylmethanesulfonyl fluoride, glycine, loading buffer, color stain protein molecular weight markers, developing and fixing kit, an enhanced chemiluminescence (ECL) color developing agent (Beyotime Biotechnology), pyroxylin or nitrocellulose (NC) membrane (Wuhan Boster Bioengineering), bovine serum albumin, acrylamide (Shanghai Boyun), anti-rat X-box binding protein 1 (XBP-I) (M-186), GADDl53(B-3), β-actin polyclonal antibody (primary antibody, US Santa Cruz), anti-rat poly (ADP-ribose) polymerase (PARP), phospho—eIF2a (Ser41) (primary antibody, US Signaling), goat anti-rabbit IgG/horseradish peroxidase (HRP; secondary antibody; Shanghai Boyun), and other reagents, such as NaF, NaCl, NaHCO3, KCl, Na2HPO4·12H2O, KH2PO4, and EDTA (analytically pure), were purchased from Jinhua Reagent Co., Ltd.
Experimental Subjects and Treatment
Experiment Subjects
The PC12 cells used in the experiment were purchased from the Institute of Neuroscience, Zhejiang University School of Medicine.
Treatment Method
The experiment was performed on the fluorine group and the control group. NaF was dissolved in water, which formed mother liquor that was 100 times. The mother liquor was sterilized under high pressure and diluted using a substrate. The cells were then treated at final concentrations of 0.5, 1.0, 1.5, and 2.0 mM [10].
Detecting Index Observation
Detecting Cell Viability and ROS
Cell viability was detected via the CCK-8 method. Cells were inoculated on a 96-well plate, with a density of 5000/100 μL. As soon as they became stable (approximately 2 h later), the cells were replaced with a medium that contained NaF and then cultivated for another 24 h. Each concentration had 12 targets, along with the cell control and the blank medium. After fluorine exposure, the cells were washed twice with phosphate-buffered saline (PBS), added into the medium with 10 % CCK-8, and incubated for 30 min. The absorption value was measured at 450 nm.
ROS were detected using the ROS assay kit. A dichlorofluorescein diacetate (DCFH-DA) probe was added. The DCFH-DA was diluted with a serum-free medium (SFM) at a ratio of 1:1000 to a final concentration of 10 μmol/L. The medium was removed, and the diluted DCFH-DA was added, incubated at 37 °C for 20 min, and washed thrice with SFM. The DCFH-DA that did not enter the cells was removed. The medium with NaF was cultivated for 2 h (the treatment period was 2 h because ROS appeared during the early stages of cell apoptosis) and then compared with the control group. Observation was conducted directly through laser scanning confocal microscopy (LSCM). The excitation wavelength was set at 488 nm, and the emission wavelength was set at 525 nm.
Observing Neuron Morphology
The neuron morphology of each group was observed via fluorescence microscopy.
Detecting Apoptosis through Hoechst-33342 Staining
After the cells were treated with NaF, the residue medium was washed with PBS, added into moderate Hoechst-33342 staining liquor to cover it, and incubated at 37 °C for 40–60 min. The staining liquor was abandoned, washed 2–3 times with PBS, and observed via LSCM. The excitation and emission wavelengths were 346 and 460 nm, respectively.
Western Blot Analysis
A separation gel (12 %, 37 °C, 1.5 h) and a concentration gel (4 %, 37 °C, l h) were formulated. Proteins were loaded to conduct electrophoresis. The constant voltage of the concentration gel was set to 80 V, whereas that of the separation gel was set to 120 V. The proteins were then transferred onto the membrane. After electrophoresis, the lengths of the proteins were determined based on the protein maker. The gel was cut out of the section where the target protein was located and then transferred onto an NC membrane. The transfer process should be performed on ice with a constant current of 300 mA for 2 h. After membrane transfer was completed, the NC membrane was placed in a confining liquid (5 % skimmed milk power with 1× TBST), vibrated gently on a shaking table at 37 °C, and confined for 2.5 h. Primary antibody incubation was conducted after confinement. The NC membrane was washed with 1× TBST to remove the milk powder and added into the primary antibody treatment fluid. PARP was diluted with XBP-1 at a ratio of 1:400, with p-elF at 1:300, and with β-actin at 1:600. Then, the mixture was incubated in a shaking table at 37 °C for 2 h and rinsed thrice with 1 × TBST for 10 min each to remove the primary antibody residue. The rinsed NC membrane was combined with goat anti-rabbit secondary antibody labeled with HRP and incubated in a shaking table at 37 °C. The diluted concentrations of the secondary antibody were 1:1000, 1:1500, and 1:3500, and the incubation durations were 1.5, 1.5, and 2 h, respectively. TBST was rinsed thrice for 10 min to remove the redundant secondary antibody. The NC membrane was placed on a glass after it was completely washed. The ECL disclosing solution was applied to cover the membrane with a preservative film. The film was exposed to develop it and then dipped in a fixing solution after a strip appeared. The film was scanned with the target protein, and semi-quantitative analysis was conducted using Quantity One software.
Statistics and Analysis
Statistical analysis was conducted using SPSS 18. Variance homogeneity testing and one-way ANOVA were adopted to conduct intergroup comparison. The least significant difference test was applied to conduct multiple comparisons with a significance standard of p < 0.05.
Result
Detecting Cell Viability with CCK-8
Cell viability was detected using the CCK-8 method, as shown in Fig. 1. Compared with that of the control group, cell viability in the 2.0 mM group decreased significantly (p < 0.05) after 2 h of fluorine exposure. The cell viability of the 1.5 and 2.0 mM groups was significantly or highly significantly decreased (p < 0.01) after 4 h of fluorine exposure. The cell viability of the 1.0 and 1.5 Mm groups decreased significantly (p < 0.05), whereas that of the 2.0 mM group decreased highly significantly (p < 0.01) after 12 h of fluorine exposure. The cell viability of the 0.5 and 1.0 Mm groups decreased significantly (p < 0.05), whereas that of the 1.5 and 2.0 mM groups decreased highly significantly (p < 0.01) after 24 h of fluorine exposure.

CCK-8 detection of cell viability
Detection Results of Intracellular ROS Concentration
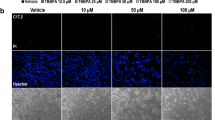
DCFH-DA was applied to detect intracellular ROS concentration. Fluorescence intensity represented intracellular ROS level, as shown in Fig. 2 and Table 1. After exposure to NaF for 2 h, the fluorescence intensity with DCF label was enhanced gradually with the incretion of fluorine concentration, which indicated that intracellular ROS increased gradually compared with the control group. The ROS levels in the 1.5 and 2.0 mM groups increased significantly (p < 0.01).

Detection of intracellular ROS
Observing Cell Morphology
The observation of the morphology results indicated that cells were typically spindle-shaped with two or more neuritides, as shown in Fig. 3. After exposure to fluorine for 24 h, cell density decreased, some of the cells turned around, apoptosis increased, and the vesicle (Fig. 3d) and cell debris (Fig. 3e) could be observed.

Cell morphology observation (10 × 20)
Detecting Apoptosis
Hoechst-33342 stain was used to detect apoptosis, as shown in Fig. 4 and Table 2. The Hoechst-33342 stain site is shown as a cell nucleus in Fig. 4. The cell nucleus is typically oblong or oval. After apoptosis, the cell nucleus became round and karyopyknosis appeared (Fig. 4e). The stain darkened, and fluorescence intensity increased. As shown in Fig. 2, no significant difference was observed in the fluorescence intensity (p > 0.05) compared with the control group. Apart from the 0.5 mM group, the other fluorine exposure groups tended to increase, which indicated that apoptosis in the fluorine exposure group exhibited the tendency to increase.

Cell apoptosis
Detection Results of PC12 Cell Protein Expression in each Group
The expression levels of the apoptosis-related proteins (p-elF, PARP) and the ERS-related protein XBP-A after 24 h of fluorine exposure are shown in Fig. 5. The results demonstrated that compared with the control group, the expression levels of ZBP-1 and PARP in the 1.0 mM group increased significantly (p < 0.05), whereas the PARP shear zone indicated that PARP expression rose significantly (p < 0.01). The concentration and protein expression levels of the other groups exhibited no significant difference (p > 0.05).

Influence of fluorine exposure on PC12 cell PARP, XBP-1, and p-elF protein expression (n = 4, \( \overline{x} \) ± s)
Discussion
Regarding the mechanism of fluorine that leads to organism damage, free radical injury theory [11–14], which is also known as oxidative stress theory, is generally recognized. Li et al. [2] indicated that oxidative stress reaction in an organism was complex and involved many factors other than SOD, MDA, and other common indices. Reddv et al. [3] reported that their research results from skeletal fluorosis patients and fluorosis rabbit indicated no significant difference in SOD, MDA, and other indices between fluorosis and the control group. To overcome the defects of traditional research index, the present study further detected intracellular ROS. A change in the redox state in cells promoted the generation of ROS and the activation of apoptosis-inducing factors, which resulted in apoptosis and aggravated change in the redox state in cells [15]. ROS detection in the current study showed that the concentration of intracellular ROS rose after 2 h of exposure to NaF as fluorine concentration increased, which indicated that intracellular ROS level was generally linearly related to fluorine exposure.
The results of this study indicate that fluorine exposure may lead to a decrease in cell viability, which is consistent with the result of the study of He Ping et al. [16], in which fluorine leads to hippocampus cell injury in primary cultured rats, as well as with our previous research results [9]. Moreover, the result of the linear relationship between cell viability and exposure time corresponds to the result of the study of Kubotap [17]. Apoptosis is a normal physiological activity that stabilizes the environment inside an organism. However, excessively high or low apoptosis is unfavorable to an organism. The present study has proven again, on the basis of the result of our previous study [9], that certain doses of NaF can lead to apoptosis and exhibit a linear relation between apoptosis level and intracellular ROS level [2, 10]. These results provide a basis to fluorine-induced free radical injury theory.
As a synthetic protein family catalyzed by poly (ADP-ribose) (PAR), PARP weighs heavily on apoptosis. Under physiological conditions, PAR uses NAD as the substrate to modify protein (mainly nucleoprotein) after translation and achieves covalent linkage of its multimer to a receptor protein involved in DNA repair to prevent cell apoptosis [18]. This protein/PAR composite consists of the DNA basic structure, several nucleoprotein polymers, and PARP from the complete DNA. Therefore, PARP can transform DNA breakage into intracellular signals, an inactive DNA repair process, or a cell death mechanism. In the caspase-dependent pathway, PARP is the substrate of caspase [19]. In vitro, PARP can be cut by various caspases, whereas in vivo, PARP is the main cutting target of caspase 3, which separates the PARP catalytic domain of c-terminus (89 kD) from the DNA binding domain of the amino terminal (24 kD), and thus, leads to enzyme activity loss. PARP is vital to cell stability and viability, which can accelerate cell instability.
The apoptosis pathway activated by ERS is called the ERS pathway, which can be divided into the following: phosphorylation of eIF-2, transcription of CHOP/Gaddl53 and activation of caspase family [20]. elF2a is an apoptosis pathway protein in apoptosis caused by ERS, although the functioning of eIF-2 is rarely reported. In general, when the unfolded protein aggregates in ER, BiP/GRP78 separates from PERK. PERK obtains a phosphorylate, forms a dipolymer, and activates the phosphorylation of elF2a to form p-eIF2a. p-elF2a cannot start a normal messenger RNA (mRNA) translation mechanism and stop protein synthesis; improperly folded protein cannot enter ER; ERS can then be released; and the ERS level is alleviated. Several studies have indicated that fluorine leads to the subunit phosphorylation of eIF2a, which decreases protein synthesis [21]. ERS can activate the cutting of XBPl mRNA, shift ORF to a code, and form a transcription factor with a leucine zipper (b-ZIP protein), which is activated XBP-1. This leucine zipper combines the ERS response element and the unfolded protein response element; it also activates all the ERS response genes, such as BiP/GRP78 and XBP-1. Unfolded protein degrades protein via weakening translation or relieves ERS through apoptosis. After 24 h of fluorine exposure, XBP-1, PARP, and the PARP shear zone of each group all share common variation tendency. Prior to fluorine exposure, concentration rises to 1.0 mM; these expression levels are proportional to exposure concentration. However, after fluorine concentration rises to 1.0 mM, a reversed ratio is observed. Compared with the control group at 1.0 mM concentration, XBP-1, PARP, and PARP cutting (PARP expression level) increase or are highly significantly increased, whereas no significant difference is observed in the expression of p-eIF2a. These findings suggest that XBP-1 and PARP are the key signaling molecules in ERS-induced apoptosis during fluorine exposure. They also suggest that during the 24-h fluorine exposure, ERS induces a hormesis effect on apoptin XBP-1 and PARP expression as well as on fluorine stain concentration. This effect is indicated by an increase in expression at low concentrations and a decrease in expression at high concentrations; that is, a parabolic relation exists between protein expression and fluorine concentration.
In conclusion, the result of this study indicates that the possible molecular mechanism of organism neuron injury during fluorine exposure may be explained as follows: excessive fluorine generates excessive ROS, induces excessive oxidative stress and ERS in the neuron, intensifies neuron apoptosis, and finally, results in injury [22, 23].
References
Shanthakumari D, Srinivasalu S, Subramanian S (2004) Effeet of fluoride intoxication on lipidperoxidation and antioxidatnt status in experimental rats. Toxicology 204(2–3):219–228
Li GS, Xu H (2005) Secondary discussion of chronical fluorosis and oxidative stress. Chin J Endemiol 24(1):3–4
Reddy GB, Khandare AL, Reddy PY, et al. (2003) Antioxidant defence system and lipid peroxidation in patients with skeletal fluorosis and in fluoride-intoxicated rabbits. Toxicol Sci 72:363
Wu Y, Zhao Q, Zhang ZG (2010) Protective effect of selenium on liver impairments induced by sodium fluoride in rats. J Environ Health 27(11):955–958
Zhao Q, Wu Y, Zhang ZG (2011) Protective effect of selenium on fluoride-induced renal impairments in rats. Chin J Endemiol 30(2):137–141
Feng P, Wei JR, Zhang ZG (2011) Intervention of selenium on chronic fluorosis-induced injury of blood antioxidant capacity in rats. Biol Trace Elem Res 144:1024–1031
Feng P, Wei JR, Zhang ZG (2012) Influence of selenium and fluoride on blood antioxidant capacity of rats. Exp Toxicol Pathol 64:565–568
Zhang L, Yang SY, Zhang ZG (2012) Intervention of selenium on liver impairment induced by chronic fluorosis in rats. J Hygiene Res 41(4):141–145
Zhang SM, Zheng XR, Zhang ZG, et al. (2015) Alterations in oxidative stress and apoptosis in cultured PC12 cells exposed to fluoride. Fluoride 48(3):213–222
Guan LY (2008) Reactive oxygen activation of endoplasmic reticulum stress and mitochondrial apoptosis pathway mediated selenite-induced apoptosis of NB4 cells. Chinese Peking Union Medical College
Xu XL, Zhang ZG, Shen XY (2001) The influence of fluorosis on learning-memory behaviors of mice and the activities of SOD and the content of MDA in the brain. China Public Health 17(1):8–10
Wang JM, Liu HT, Ma JJ (2006) Fluorosis and application of interrelated technology in it. Prog Vet Med 27(3):51–54
Hefnawy G, Pérez T (2010) The importance of selenium and the effects of its deflciency in animal health. Small Ruminant Res 89:185–192
Barbier O, Mendoza LA, del Razo LM (2010) Molecular mechanisms of fluoride toxicity. Chem Biol Interact 188:319–333
Zhou YT, Xiao HB, Bi MG (2011) ROS and endoplasmic reticulum stress. Chinese Pharmacological Bulletin 27(5):597–600
He P, Zhang M, He WH (2006) Effects of fluoride on oxidative stress and apoptosis in primary rat hippocampal neurons.chinese. J Endemiol 25(3):264–267
Kubota K, Lee DH, Tsuchiya M (2005) Fluoride induces endoplasmic reticulum stress in ameloblasts responsible for dental enamel formation. J Biol Chem 280(24):23194–23202
Zhang QX, Zhao H, Zhang C (2013) Wind, tonify deficiency medicine caspase 3 on cerebral ischemia rats and the effects of PARP expression. J Beijing Univ Chin Med 36(4):246–249
Song XY, Hu JF, Chen NH (2012) Neural cell apoptosis with cerebral ischemia disease. Chin Pharmacol 28(3):307–310
Feng LJ, Wang HP, et al. (2007) Endoplasmic reticulum stress and the degeneration of neurons. Chinese Pharmacology 23(4):428–432
Sharma R et al. (2008) Fluoride induces endoplasmic reticulum stress and inhibits protein synthesis and secretion. Environ Health Perspect 9(116):1142–1146
Li GS (2003) Calcium from the “contradiction” look at the mechanism of skeletal fluorosis. National conference on fluorine and arsenic, Harbin: Chinese journal of endemiology newsroom
Xiong XZ, Liu JL, He WH, et al. (2007) Dose-effect relationship between drinking warer fluoride levels and damage to liver and kidney functions in children. Environ Mental Res 103(1):112–116
Acknowledgments
This research was sponsored by the National Natural Science Foundation of China, grant number: 81273015, and the National Natural Science Foundation of China, grant number: 81573101.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ke, L., Zheng, X., Sun, Y. et al. Effects of Sodium Fluoride on Lipid Peroxidation and PARP, XBP-1 Expression in PC12 Cell. Biol Trace Elem Res 173, 161–167 (2016). https://doi.org/10.1007/s12011-016-0641-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-016-0641-3