
Abstract
Quinolinic acid (QUIN) striatal injection in rat reproduces the main neurochemical features of Huntington’s disease (HD), including oxidative damage. In this study, we evaluated the effect of a copper (Cu) supplement in drinking water (90 ppm Cu, 28 days) on the QUIN-induced HD model in the rat. Copper exposure caused no signs of liver toxicity; however, it produced significant Cu accumulation in striatum. It is noteworthy that QUIN also caused increased striatal Cu content; when the supplement was administered to animals with QUIN-injury, an even higher metal striatal accumulation was observed. Cu pre-treatment preserved striatal gamma-aminobutyric acid (GABA) content, which was reduced by QUIN intrastriatal injection. Similarly, apomorphine-induced circling behavior was reduced in Cu-pretreated QUIN-damaged rats. Metal supplement in drinking water prevented both lipid peroxidation and reactive oxygen species (ROS) formation caused by QUIN in striatum. In Cu-treated groups, superoxide dismutase-1 (SOD1) activity showed a significant increase, while SOD2 activity was slightly enhanced. Although the pathophysiological role for higher Cu levels in patients with HD and in experimental models of the disease is not fully understood, results in the present study suggest that Cu oral intake stimulates anti-oxidant defenses, an effect that may be a potential factor for reducing the progression of HD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Huntington’s disease (HD) is a neurodegenerative disorder caused by a mutation in the gene that encodes the huntingtin protein (350 kDa) [1–3]. Small huntingtin N-terminal fragments have been implicated as the main mediators of disease progression [4]. These fragments possess aberrant interactions among themselves as well as with other biomolecules, which possibly result in the HD mechanism of damage, including protein aggregation [5], transcriptional repression [6], altered tryptophan metabolism [7], oxidative injury [8, 9], and mitochondrial dysfunction [10]. Notably, increased content of copper (Cu) and iron (Fe) in the caudate–putamen from postmortem HD brain has been observed [11]; however, the specific role of Cu in HD set up or progression remains unknown.
Physiologically, Cu plays an important role in endogenous antioxidant proteins and in neurotransmitter synthesis [12]. In the central nervous system (CNS), Cu levels are related to Fe concentration by specific metal transporters such as Cu transporter-1, divalent metal transporter-1, antioxidant protein-1, and ATP7A [13]. A significant increase in striatal Cu has been reported after the administration of a single unilateral, intrastriatal injection of quinolinic acid (QUIN) to rats, a model of HD [14]. QUIN causes neuronal death by oxidative stress (OS), which is accompanied by gamma aminobutyric acid (GABA) depletion [15–18]. Both in vivo and in vitro studies have shown that Cu negatively modulates N-methyl-D-aspartate (NMDA) receptors [19–21]. Following this evidence, Santamaría et al. demonstrated the neuroprotective effect of systemic Cu administration in rats subjected to a single unilateral, intrastriatal QUIN injection [22]. However, other studies suggest that increased amounts of Cu, bound to low-affinity sites, may contribute to pro-oxidant activities and even neurodegeneration [23]; therefore, the Cu neuroprotective effect may continue to be debated.
The role of Cu in the set up and/or development of HD remain uncertain; thus, in this work, we supplemented rats with Cu to observe whether this metal exerts neuroprotection by modulating OS.
Materials and Methods
Biological Material
Male Wistar rats weighing 200–250 g at the beginning of the experiments were used throughout the study. All animal procedures were approved by the Institutional Review Committee and were in accordance with the current Mexican Legislation NOM-062-ZOO-1999 norm and in agreement with National Institutes of Health (NIH, Bethesda, MD, USA) guide criteria for the care and use of laboratory animals. The animal housing room was maintained under constant temperature (25 ± 3 °C), humidity (50 ± 10 %), and lighting (12-h:12-h light to dark cycles).
Experimental Design
Animals were randomly assigned to one of two protocols (Fig. 1). Protocol I: 72 rats were divided into 2 groups, one group received regular drinking water, and the second received 90 parts per million (ppm) of Cu solution (Copper sulfate [CuSO4]•5H2O, 350 mg/L) for 28 days. This concentration of Cu has been utilized in nutritional studies as Cu supplement for rodents [24, 25]. Both drinking water and Cu solution were replaced on a daily basis, and the volume of liquid ingested was recorded and used to estimate individual intake. Animal body weight (BW) was also recorded. After 28 days of exposure to the metal, each group was further divided into two subgroups: one subgroup of each treatment received a QUIN (240 nmol/μL) intrastriatal injection of 1 μL, and the remaining subgroup received an equivalent volume of saline solution (SS). The intrastriatal injection was performed under deep anesthesia with sodium pentobarbital [40 mg/kg, intraperitoneal (i.p.)] and the stereotaxic coordinates were as follows: 0.5 mm anterior, 2.7 lateral to bregma, and 4.5 ventral to dura [26]. Two hours later, blood samples were collected, and immediately after, rats were sacrificed. Striata were dissected and stored at −85 °C for lipid peroxidation (LP), reactive oxygen species (ROS) measurement, superoxide dismutase (SOD) activities, and Cu determination. For this late assay, samples were collected into sterile polypropylene Cu-free tubes. Protocol II: Twenty-four rats were used in this study. They were treated similar to Protocol I but, once the right striatal lesion has occurred, they were allowed 6 days to develop the neurochemical characteristics of HD. On day 6th, circling behavior was evaluated and 1 day later rats were sacrificed to obtain striata. Samples were stored at −85 °C and GABA content was determined [17].

Experimental design. The figure shows a schematic representation of experimental protocol I and II followed in this study, experimental groups, the time frame, and analytical determinations. Number of animals used is indicated per group. Quinolinic acid (QUIN) (240 nmol/μL), Saline solution (SS) solution, Blood sample (BS), Sacrifice (Sac), Circling behavior (CB), Gamma aminobutyric acid (GABA)
Determination of Striatal Cu
Striatal Cu content was determined by atomic absorption spectrophotometry (Perkin-Elmer 3110 Atomic Absorption Spectrophotometer equipped with an HGA-600 graphite furnace and an AS-60 autosampler). Tissue samples were digested using 1 mL concentrated metal-free nitric acid (Suprapur, Merck, Mexico) in a water bath at 60 °C for 30 min. After digestion, aliquots of the solution were then injected into the graphite furnace for Cu analysis. Calibration curves were constructed by using commercial standard Cu (Titrisol Merck, Mexico). An external biological standard from National Institute of Standards and Techniques (NIST; bovine liver cat. no. 1577b) was analyzed in every session and was considered valid only if the results from this analysis were between 95 % and 105 % of those in the certificate of analysis. Results are expressed as μg Cu/g wet tissue [27].
Assessment of Liver Toxicity
The plasma activities of Gamma-glutamyl transpeptidase (GGT) and Alanine transaminase (ALT) were measured as markers of liver toxicity. After 28 days of Cu treatment, rats were anesthetized with sodium pentobarbital (45 mg/kg) and blood samples were obtained by cardiac puncture; plasma was obtained by centrifugation and employed to determine enzymatic activities [28, 29].
Circling Behavior Evaluation
Circling behavior was performed 6 days after QUIN injury, this period is necessary in order to assess the imbalance in dopamine receptors caused by the unilateral injection of QUIN; the number of turns is proportional to the extent of damage caused [17, 22]. Animals were dosed with apomorphine [1 mg/kg, subcutaneous (s.c.)] and 5 min later, each rat was placed in an individual acrylic cage and the number of ipsilateral turns was recorded during 60 min. Circling behavior evaluation was carried out by a researcher blinded to the treatments.
Determination of Striatal GABA
The striatal GABA content was assayed by high-performance liquid chromatography (HPLC) coupled with a fluorescence detector, as previously described [30]. On day 7 after QUIN injection (the day following that of the circling behavior evaluation), rats were injected in the caudal vein (i.v.) with 3-mercaptopropionic acid (1.2 mmol/kg), a glutamate decarboxylase (GAD) inhibitor, to prevent post mortem changes in GABA levels, and 1 min later, the animals were sacrificed and the striatum was obtained. Samples were stored at −85 °C until analysis. The striatum tissue was homogenized in 1 mL of 85 % HPLC-grade methanol by using an ultrasonic processor (130 W, 40 % amplitude, 10–15 s); samples were then centrifuged at 18,500 rpm, 15 min, 4 °C, and supernatants were pre-column-derivatized with ortho-phthalaldehyde reagent prior to analysis.
Determination of Striatal Oxidative Damage
Lipid Peroxidation
The formation of lipid-soluble fluorescent compounds in striatum was measured according to Martínez-Lazcano et al. [31]. The lesioned striatum was homogenized in 3 mL of saline solution (SS); 1 mL of this homogenate was added to 4 mL of a chloroform-methanol (2:1 v/v) mixture. Samples were then vortex-mixed and placed on ice for 30 min in the dark; after this time, the upper phase was removed and the fluorescence in the organic layer was determined in a spectrofluorometer at 350-nm excitation and 430-nm emission wavelengths. Results of fluorescence were normalized by protein content. Results are expressed as relative fluorescence units per mg of protein (RFU/mg protein).
Reactive Oxygen Species
ROS were evaluated in 5 μL of striatal homogenate according to a previously reported method modified for brain tissue [31]. A fluorometric assay based on the oxidation of 2′7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Molecular Probes, USA) into dichlorofluorescein (DCF), was employed. Results are expressed as nmol DCF/mg prot/min.
Determination of Cu–Zn SOD and Mn SOD Activities
The lesioned striatum was homogenized in phosphate buffer (50 mM, pH 7.4), and 20 μL of the homogenate were incubated with 2.9 mL of a solution containing 10 μM NaN3, 10 μM reduced cytochrome c, 10 mM disodium ethylenediaminetetraacetic acid (EDTA), 100 μM xanthine, 20 mM sodium bicarbonate, and 0.02 % Triton X-100, pH 10.2. The reaction was started by the addition of 50 μL of xanthine oxidase (3.4 mg/mL in 0.1 mM EDTA). Change in absorbance was monitored every 30 s at 550 nm. The participation of Cu–Zn SOD was calculated as total activity minus activity measured in the presence of potassium cyanide (KCN; 1 mM). One unit of SOD activity was defined as the amount of enzyme that decreased the reduction rate of cytochrome c by 50 % [32].
Statistical Analysis
Data are expressed as mean ± standard error of the mean (SEM). Mean differences between control and Cu-exposed animals in water intake, BW, and liver toxicity were analyzed by Student t test. For comparison of circling behavior, the Kruskal–Wallis test followed by the Mann–Whitney U was used. Differences in Cu content, oxidative parameters, and enzymatic activities were analyzed by one-way analysis of variance (ANOVA) followed by Tukey test. In all the cases, significance was considered when p < 0.05. The R ver. 2.11.1 software package was employed.
Results
Cu Supplement in Drinking Water
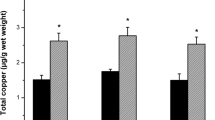
Cu supplement for 28 days, at the concentration assayed, caused no effect on the animals’ food intake, BW, or drinking water intake as compared with controls (data not shown). The supplement of Cu in drinking water increased striatal metal levels (4.35 ± 0.1193 μg/g tissue) when compared to controls (2.45 ± 0.317 μg/g tissue). It is noteworthy that QUIN-induced injury by itself also caused a statistical increase in striatal Cu (5.13 ± 0.09 μg/g tissue). We also observed that Cu pretreatment in QUIN-injured animals further increased striatal Cu (7.54 ± 0.76 μg/g tissue) compared with animals with QUIN intrastriatal injection alone or with those with metal supplement without injury (F 3,20 = 27.95; p <0.0001; Fig. 2).

Striatal Cu content. Rats were supplemented with 90 parts per million (ppm) Cu in drinking water for 28 days (controls received regular tap water). Then, the animals were infused with either 240 nmol/μL of quinolinic acid (QUIN) or saline solution (SS). Two hours later, animals were sacrificed and striatum obtained to determine Cu by atomic absorption spectrophotometry. *p < 0.05; **p < 0.01; *p < 0.001, differences from control tap water/SS; + p < 0.01, different from Cu sulfate (CuSO4)/SS; # p < 0.01 different from tap water/QUIN. Analysis of variance (ANOVA) followed by Tukey test (n = 6 for each group)
Effect of Sub-Chronic Cu Supplement on Liver Function
The activity of the enzymes ALT (marker of hepatocyte necrosis) and GGT (associated with hepatocyte membrane damage) in control animals or in those supplemented with 90 ppm Cu in drinking water for 28 days are depicted in Table 1. No statistical difference between groups was observed (t 2.447,2,6 = 1.886, p = 0.1082 for ALT and t 2.447,2,6 = 1.718; p = 0.1609 for GGT).
Effect of Cu Treatment and QUIN-Induced Injury on Circling Behavior and Striatal GABA Levels
Six days after striatal SS infusion, the animals showed no signs of brain damage, evidenced as a lack of turning in response to apomorphine challenge. As expected, rats with QUIN-induced striatal lesion exhibited turning behavior presenting about 300 turns, ipsilateral to the lesioned side, after a dose of the dopaminergic agonist, as a consequence of the imbalance in dopamine receptors between hemispheres caused by the injury. Interestingly, QUIN-lesioned rats previously supplemented with Cu showed a significant reduction (KW2 = 13.14; p = 0.001) in the number of turns (79 ± 20) compared with rats with QUIN-induced striatal lesion without Cu supplement (Fig. 3a).

Effect of Cu supplement and quinolinic acid (QUIN) in a) apomorphine-induced circling behavior and b) gamma-aminobutyric acid (GABA) level determination. Rats were supplemented with 90 parts per million (ppm) Cu in drinking water for 28 days (controls received regular tap water). Then, animals were infused with either 240 nmol/μL QUIN or saline solution (SS). Animals were evaluated for circling behavior 6 days after QUIN injury, and GABA was determined 7 days after QUIN infusion. ***p < 0.001 differences from control tap water/SS; + p < 0.01 differences from CuSO4/SS; # p < 0.01 differences from Cu sulfate (CuSO4)/QUIN (n = 6 for each group). Kruskal–Wallis followed by Mann–Whitney U test for circling behavior, and analysis of variance (ANOVA) followed by Tukey test for GABA levels was tested
Regarding striatal GABA, control rats showed 2.74 ± 0.06 nmol/mg of tissue. QUIN intrastriatal injection statistically reduced GABA content (0.89 ± 0.17 nmol/mg tissue; p < 0.001). Interestingly, Cu supplement in drinking water prevented QUIN-induced GABA decrease (2.01 ± 0.36 nmol/mg tissue; F 3,20 = 25.51; p = 0.0002; Fig. 3b).
Cu Prevents QUIN-Induced Striatal Oxidative Damage
The OS status was evaluated as the formation of ROS and LP in the injured striatum. Results showed that QUIN induced significant ROS formation (9.34 ± 0.24 nmol DCF/mg prot/min) as compared with the control group (5.61 ± 0.44 nmol DCF/mg prot/min). Cu supplementation partially prevented the ROS formation (7.22 ± 0.55 nmol DCF/mg prot/min) caused by QUIN (F 3,20 = 9.254; p = 0.001; Fig. 4a). Results in LP exhibited consistence with those of the ROS assay. QUIN-induced striatal injury produced a significant increase of fluorescent lipids derived from oxidative damage (5.38 ± 0.48 RFU/mg prot) as compared with the control group (1.85 ± 0.15 RFU/mg prot). Cu supplement enabled partial protection against the oxidative damage induced by QUIN (3.10 ± 0.31 RFU/mg prot; F 3,20 = 25.39; p < 0.0001; Fig. 4b).

Effect of Cu supplement on quinolinic acid (QUIN)-induced. a) Reactive oxygen species (ROS) formation, and b) Lipid peroxidation in rats. Cu supplement was given in the drinking water for 28 days; then, the animals were infused with QUIN in the striatum and 2 h later, rats were sacrificed and the striatal tissue was obtained to assess ROS generation and the content of the fluorescent lipid derived from oxidative stress. *p < 0.05, **p < 0.01, and ***p < 0.001 different from control tap water/saline solution (SS); + p < 0.01 differences from Cu sulfate (CuSO4)/SS; # p < 0.01 differences from CuSO4/QUIN (n = 6 for each group). Analysis of variance (ANOVA) followed by Tukey test
Effect of Cu Pretreatment on Cu–Zn SOD and Mn SOD Activities
Cu–Zn SOD and Mn SOD activities in striata were evaluated 2 h after QUIN lesion (Fig. 5). Infusion of QUIN produced a 27 % reduction in Cu–Zn SOD activity (0.185 ± 0.008 enzyme units/mg prot) as compared with the control group (0.2570 ± 0.006 enzyme units/mg prot), while pretreatment with the Cu supplement not only reverted this effect, but increased enzyme activity twofold compared with controls (0.4997 + 0.0525 enzyme units/mg prot). Enhanced Cu–Zn SOD activity was only observed in tissue from animals with Cu supplement and injured with the toxin; whereas in animals with Cu supplement without the lesion, no statistical increase in enzyme activity was observed (F 3,20 = 25.32; p = 0.0002; Fig. 5a). Regarding the Mn SOD isoform, we observed that the activity of this enzyme was increased by QUIN-induced injury (0.3080 enzyme units ± 0.0032) as compared with the control group (0.1293 ± 0.008 enzyme units/mg prot). In this context, Cu supplement further increased Mn SOD activity in rats with injury (0.2402 ± 0.02 enzyme units/mg prot) without reaching statistical significance (F 3,20 = 8.223; p = 0.0025; Fig. 5b).

Effect of Cu supplement 90 parts per million (ppm) in drinking water for 28 days and quinolinic acid (QUIN) striatal infusion on superoxide dismutase (SOD) activity in rats. a) Cu–Zinc-dependent SOD isoform (Cu–Zn SOD), and b) manganese-dependent SOD isoform (Mn SOD). *p < 0.05; **p < 0.01 different from control tap water/saline solution (SS); + p < 0.01 differences from Cu sulfate (CuSO4)/QUIN; # p < 0.01 differences from tap water/QUIN (n = 6 for each group). Analysis of variance (ANOVA) followed by Tukey test
Discussion
Copper is found in mammals as a trace element and is essential for the structure and function of several enzymes [12]. In the CNS, Cu participates as co-factor of antioxidant enzymes such as ceruloplasmin and Cu–Zn SOD. However, some reports associate Cu with free radical-mediated toxic processes such as the Fenton reaction [9]. The apparent duality of Cu has prompted research groups to study its participation in neurodegenerative processes. Likewise, Dexter et al. studied postmortem brain tissue from patients with HD and found Cu at higher concentrations than in controls [11]. The same finding has been observed in HD experimental models involving excitotoxicity [14, 23], suggesting that Cu could play a role at the onset of neuronal damage in HD. In contrast, other research groups have demonstrated that Cu exerts a neuroprotective effect in HD, Parkinson’s disease (PD), and Alzheimer’s disease (AD) experimental models regarding excitotoxicity and cell death [17, 24, 33, 34]. In a previous work, our group showed that Cu administered i.p. showed protection against QUIN striatal injection; however, the administration pathway used in that study is not relevant for an essential trace metal [17].
To determine whether Cu in drinking water at 90 ppm for 28 days promoted liver toxicity, we evaluated serum ALT and GGT activities. We observed only a slight, non-significant increase in serum ALT, and no effect on GGT; therefore, we suggest that Cu supplement at the concentration and time of exposure used in the present study was safe for rats. However, the Cu concentration in drinking water favored the entry of this metal into the brain, because supplemented rats doubled the brain Cu content of that of controls. We believe that the oral sub-toxic Cu concentration employed in the present study was subjected to physiological regulation of intestinal absorption and to hepatic homeostasis [35–37]. Therefore, at the scheme utilized here, the Cu supplement was adequate for favoring the entry of the metal into the brain without causing significant liver damage.
It is noteworthy that, in the present study, animals subjected to striatal QUIN-induced injury without metal supplement showed increased Cu content in comparison with controls (Fig. 2), a finding that can be related with the fact that post mortem brains from patients with HD at the initial disease stage exhibited increased Cu levels in putamen and substantia nigra pars compacta, compared with controls [9]. The same effect was observed in QUIN-induced HD experimental rats, in which a higher striatal Cu content was demonstrated [14, 22]. The effect observed herein may be due to the fact that Cu transport is linked with glutamate neurotransmission; in some brain regions, it has been observed that Cu is released after activation of NMDA-type glutamate receptors. Furthermore, stimulation of those receptors results in the trafficking of ATP7A, a Cu-specific transporter, out of the late Golgi into dendrites [38–40], a phenomenon leading to Cu accumulation in the stimulated area. This could be the case for QUIN-mediated overstimulation of striatal NMDA-type glutamate receptors. This possibility is consistent with the fact that a previous load of Cu in the rat, by Cu supplementation, increased the availability of the metal that, after the noxious stimulus resulted in a much higher accumulation of Cu than in either Cu-exposed animals but not in QUIN-damaged, or in QUIN-damaged but not in Cu-supplemented. Other authors have related increased Cu levels with mechanisms of cell damage in HD [11, 23]; however, it is also possible that the increased content of this transition metal in the basal ganglia could be part of a defense mechanism [20, 21]. Marchetti et al, showed that in vitro exposure to Cu (≤30 mM) can facilitate the NMDA receptor current, but higher concentration of Cu can inhibit the activity of the receptor. Therefore, the concentration of Cu in the brain is highly relevant to determinate its activity at synaptic sites and the fate of the cell [41]. The results from the present study are in agreement with the latter possibility, because we observed a preservation of GABA in Cu-supplemented animals, along with a decreased number of apomorphine-elicited turns than in the QUIN-injured group; both findings suggest that the Cu load in these animals provided partial protection to GABA-producing neurons against damage caused by QUIN.
We consider that the protection conferred by Cu could be related with its ability to induce a higher participation of antioxidant proteins. In this regard, SODs comprise a group of key enzymes involved in antioxidant defense. The Cu–Zn-dependent isoform, SOD1, participates detoxifying intracellular free radicals and protecting cells by the conversion of superoxide anion into hydrogen peroxide [42]. The deficiency of SOD activity or expression had been related with several pathologies [27, 43, 44]. Boll et al. in 2008 reported that in patients with HD, SOD1 activity is lower as compared with that of controls, as well as Cu-dependent ferroxidase ceruloplasmin activity [27]. In our study, Cu–Zn SOD activity was increased in striata from Cu-pretreated QUIN-injured rats. A possible explanation for this is an increase in Cu–Zn SOD protein content. Previous reports show that rats acutely exposed to Cu exhibited higher striatal Cu–Zn SOD content [17]. Therefore, it is reasonable to speculate that longer (sub-chronic) exposure to Cu may also increase this protein content. Additionally, the role of free Cu as anion superoxide scavenger cannot be underestimated [45]. Our data also support those of Barik [45] and could explain Cu-enabled protection. Another possibility for the neuroprotective effect of Cu involves its ability to inhibit NMDA receptor activity. Electrophysiological studies showed that Cu ions are able to inhibit NMDA receptors in hippocampus and cortex cultures, and Cu was further described as high-affinity antagonist for NMDA, independently from receptor activation [20, 21]. Furthermore, Cu at the micromolar range is able to antagonize NMDA and GABA receptors in isolated cells and olfactory bulb interneurons [19]. Another important issue to consider regarding Cu is its ability to inhibit the effects of nitric oxide (NO) [46]. This is relevant because it was demonstrated that in HD transgenic mice, the activity of neuronal nitric oxide synthase (nNOS) is increased [47], which positively correlates with an increase in NO production and the consequent oxidative-nitrosative damage.
The World Health Organization in the Guidelines for Drinking-water Quality established a provisional guideline value of 2 mg/L to be protective against the adverse effects of copper and to provide an adequate margin of safety in populations with normal copper homeostasis [48]. However, differences in human Cu ingestion allowances exist. In this regard, Pal et al. have recently reviewed the range of safety for human Cu ingestion and it fluctuates from few μg/L to 10 mg/L [49]. Although the plain numbers comparisons (2 mg/L, for humans vs. 90 mg/L, for rats) seems quite different, there are considerations to be made. A mean drinking water volume for a 250-g rat is 30 mL, while human average consumption is 2.5 L. Given this, the daily safe amount of Cu ingestion for humans would be 5 mg, while the amount of Cu administered daily to the rats was 2.7 mg. It is relevant to mention that substantial differences in terms of absorption, distribution, and metabolism, among others, exist between the two species. Therefore, a suggestion of lineal extrapolation of Cu dose from rats to humans would require several clinical considerations as well as the evaluation of biochemical markers as suggested by Pal et al. [50].
Our results suggest that Cu supplement in drinking water conferred protection, assessed as GABA preservation and decreased turning behavior, against the effects caused by the NMDA-receptors agonist QUIN. Copper treatment favored SOD activity and limited LP; therefore, induction of the endogenous antioxidant system may be involved. The increased Cu content found in basal ganglia of HD patients and in experimental models of the disease could be a compensatory mechanism to diminish free radicals; undoubtedly, more studies are necessary to fully elucidate the role of copper in HD.
References
MacDonald ME, Ambrose CM, Duyao MP et al (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72:971–983
Tobin AJ, Signer ER (2000) Huntington’s disease: the challenge for cell biologists. Trends Cell Biol 10:531–536
Vonsattel JP, Myers RH, Stevens TJ et al (1985) Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 44:559–577
Graham RK, Deng Y, Slow EJ et al (2006) Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125:1179–1191
DiFiglia M, Sapp E, Chase KO et al (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993
Dunah AW, Jeong H, Griffin A et al (2002) Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science 296:2238–2243
Browne SE, Bowling AC, MacGarvey U et al (1997) Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 4:646–653
Stoy N, Mackay GM, Forrest CM et al (2005) Tryptophan metabolism and oxidative stress in patients with Huntington’s disease. J Neurochem 93:611–623
Gines S, Ivanova E, Seong IS et al (2003) Enhanced Akt signaling is an early pro-survival response that reflects N-methyl-D-aspartate receptor activation in Huntington’s disease knock-in striatal cells. J Biol Chem 278:50514–50522
Seong IS, Ivanova E, Lee JM et al (2005) HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Genet 14(287):1–80
Dexter DT, Carayon A, Javoy-Agid F et al (1991) Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114:1953–1975
Madsen E, Gitlin JD (2007) Copper and iron disorders of the brain. Annu Rev Neurosci 30:317–337
Monnot AD, Zheng G, Zheng W (2012) Mechanism of copper transport at the blood-cerebrospinal fluid barrier: influence of iron deficiency in an in vitro model. Exp Biol Med 237:327–333
Perez P, Flores A, Santamaria A et al (1996) Changes in transition metal contents in rat brain regions after in vivo quinolinate intrastriatal administration. Arch Med Res 27:449–452
El-Defrawy SR, Boegman RJ, Jhamandas K, Beninger RJ (1986) The neurotoxic actions of quinolinic acid in the central nervous system. Can J Physiol Pharmacol 4:369–375
Kalonia H, Kumar P, Kumar A (2011) Comparative neuroprotective profile of statins in quinolinic acid induced neurotoxicity in rats. Behav Brain Res 216:220–228
Santamaria A, Flores-Escartin A, Martinez JC et al (2003) Copper blocks quinolinic acid neurotoxicity in rats: contribution of antioxidant systems. Free Radic Biol Med 35:418–427
Stipek S, Stastny F, Platenik J et al (1997) The effect of quinolinate on rat brain lipid peroxidation is dependent on iron. Neurochem Int 30:233–237
Trombley PQ, Shepherd GM (1996) Differential modulation by zinc and copper of amino acid receptors from rat olfactory bulb neurons. J Neurophysiol 76:2536–2546
Vlachova V, Zemkova H, Vyklicky L (1996) Copper modulation of NMDA responses in mouse and rat cultured hippocampal neurons. Eur J Neurosci 8:2257–2264
Weiser T, Wienrich M (1996) The effects of copper ions on glutamate receptors in cultured rat cortical neurons. Brain Res 742:211–218
Santamaria A, Rios C, Perez P et al (1996) Quinolinic acid neurotoxicity: in vivo increased copper and manganese content in rat corpus striatum after quinolinate intrastriatal injection. Toxicol Lett 87:113–119
Fox JH, Kama JA, Lieberman G et al (2007) Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One 2:e334
Alcaraz-Zubeldia M, Rojas P, Boll C, Ríos C (2001) Neuroprotective effect of acute and chronic administration of copper (II) sulfate against MPP + neurotoxicity in mice. Neurochem Res 26:59–64
Crowe A, Morgan EH (1996) Iron and copper interact during their uptake and deposition in the brain and other organs of developing rats exposed to dietary excess of the two metals. J Nutr 126:183–194
Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates, 2nd edn. Academic Press, San Diego, CA, USA
Boll MC, Alcaraz-Zubeldia M, Montes S, Ríos C (2008) Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NO(x) content in the CSF. A different marker profile in four neurodegenerative diseases. Neurochem Res 33:1717–1723
Glossmann H, Neville DM (1972) Gamma-glutamyltransferase in kidney brush border membranes. FEBS Lett 19:340–344
Reitman S, Frankel S (1957) A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am J Clin Pathol 28:56–63
Perez-Neri I, Castro E, Montes S et al (2007) Arginine, citrulline and nitrate concentrations in the cerebrospinal fluid from patients with acute hydrocephalus. J Chromatogr B Analyt Technol Biomed Life Sci 851:250–256
Martínez-Lazcano JC, Perez-Severiano F, Escalante B et al (2007) Selective protection against oxidative damage in brain of mice with a targeted disruption of the neuronal nitric oxide synthase gene. J Neurosci Res 85:1391–1402
Crapo JD, McCord JM, Fridovich I (1978) Preparation and assay of superoxide dismutases. Methods Enzymol 53:382–393
Acevedo KM, Hung YH, Dalziel AH et al (2011) Copper promotes the trafficking of the amyloid precursor protein. J Biol Chem 286:8252–8262
Scheiber IF, Mercer JF, Dringen R (2014) Metabolism and functions of copper in brain. Prog Neurobiol 116:33–57
Linder MC, Hazegh-Azam M (1996) Copper biochemistry and molecular biology. Am J Clin Nutr 63:797S–811S
Turnlund JR, Keyes WR, Anderson HL, Acord LL (1989) Copper absorption and retention in young men at three levels of dietary copper by use of the stable isotope 65Cu. Am J Clin Nutr 49:870–878
Zhang Y, Li B, Chen C, Gao Z (2009) Hepatic distribution of iron, copper, zinc and cadmium-containing proteins in normal and iron overload mice. Biometals 22:251–259
Dodani SC, Domaille DW, Nam CI et al (2011) Calcium-dependent copper redistributions in neuronal cells revealed by a fluorescent copper sensor and X-ray fluorescence microscopy. Proc Natl Acad Sci USA 108:5980–5985
Gaier ED, Eipper BA, Mains RE (2013) Copper signaling in the mammalian nervous system: synaptic effects. J Neurosci Res 91:2–19
Schlief ML, West T, Craig AM et al (2006) Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc Natl Acad Sci USA 103:14919–14924
Marchetti C, Baranowska-Bosiacka I, Gavazzo P (2014) Multiple effects of copper on NMDA receptor currents. Brain Res 1542:20–31
McCord JM, Fridovich I (1969) Superoxide dismutase. J Biol Chem 244:6049–6055
Johnson F, Giulivi C (2005) Superoxide dismutases and their impact upon human health. Mol Aspects Med 26:340–352
Uchino M, Ando Y, Tanaka Y et al (1994) Decrease in Cu/Zn- and Mn-superoxide dismutase activities in brain and spinal cord of patients with amyotrophic lateral sclerosis. J Neurol Sci 127:61–67
Barik A, Mishra B, Shen L, Mohan H, Kadam RM, Dutta S, Zhang HY, Priyadarsini KI (2005) Evaluation of a new copper(II)-curcumin complex as superoxide dismutase mimic and its free radical reactions. Free Radic Biol Med 39(6):811–822
Plane F, Wigmore S, Angelini GD, Jeremy JY (1997) Effect of copper on nitric oxide synthase and guanylyl cyclase activity in the rat isolated aorta. Br J Pharmacol 121:345–350
Perez-Severiano F, Escalante B, Vergara P et al (2002) Age-dependent changes in nitric oxide synthase activity and protein expression in striata of mice transgenic for the Huntington’s disease mutation. Brain Res 951:36–42
WHO. (2004) Copper in drinking-water background document for development of WHO guidelines for drinking-water quality http://www.who.int/water_sanitation_health/dwq/chemicals/copper.pdf
Pal A, Jayamani J, Prasad R (2014) An urgent need to reassess the safe levels of copper in the drinking water: lessons from studies on healthy animals harboring no genetic deficits. Neurotoxicology 44:58–60
Pal A (2014) Copper toxicity induced hepatocerebral and neurodegenerative diseases: an urgent need prognostic biomarkers. Neurotoxicology 40:97–101
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Martínez-Lazcano, J.C., Montes, S., Sánchez-Mendoza, M.A. et al. Sub-Chronic Copper Pretreatment Reduces Oxidative Damage in an Experimental Huntington’s Disease Model. Biol Trace Elem Res 162, 211–218 (2014). https://doi.org/10.1007/s12011-014-0127-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-014-0127-0