
Abstract
Chromium picolinate (CrPic) has been indicated to activate glucose transporter 4 (GLUT4) trafficking to the plasma membrane (PM) to enhance glucose uptake in 3T3-L1 adipocytes. In skeletal and heart muscle cells, insulin directs the intracellular trafficking of the fatty acid translocase/CD36 to induce the uptake of cellular long-chain fatty acid (LCFA). The current study describes the effects of CrPic and insulin on the translocation of CD36 from intracellular storage pools to the PM in 3T3-L1 adipocytes in comparison with that of GLUT4. Immunofluorescence microscopy and immunoblotting revealed that both CD36 and GLUT4 were expressed and primarily located intracellularly in 3T3-L1 adipocytes. Upon insulin or CrPic stimulation, PM expression of CD36 increased in a similar manner as that for GLUT4; the CrPic-stimulated PM expression was less strong than that of insulin. The increase in PM localization for these two proteins by insulin paralleled LCFA ([1-14C]palmitate) or [3H]deoxyglucose uptake in 3T3-L1 adipocytes. The induction of the PM expression of GLUT4, but not CD36, or substrate uptake by insulin and CrPic appears to be additive in adipocytes. Furthermore, wortmannin completely inhibited the insulin-stimulated translocation of GLUT4 or CD36 and prevented the increased uptake of glucose or LCFA in these cells. Taken together, for the first time, these findings suggest that both insulin and CrPic induce CD36 translocation to the PM in 3T3-L1 adipocytes and that their translocation-inducing effects are not additive. The signaling pathway inducing the translocations is different, apparently resulting in a differential activity of CD36.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Long-chain fatty acid (LCFA) is one of the major substrates from which energy is generated to sustain life processes [1]. In mammalian cells, uptake of LCFA occurs by two different processes: (1) passive diffusion through the phospholipid bilayer of the plasma membrane (PM) and (2) facilitated transport across the PM mediated by one or more specific membrane proteins [2, 3]. In the latter process, different types of LCFA transporter proteins have been identified, namely, fatty acid translocase (FAT)/CD36 [4], PM-bound fatty acid binding protein (FABPpm) [5, 6], and the fatty acid transporter protein family (FATP1-6) [7, 8]. CD36 is a multifunctional membrane glycoprotein that has been extensively studied and identified to be a key determinant in cellular FA uptake [9]. When overexpressed in transgenic mice, CD36 increases FA uptake [10]. Moreover, CD36 knockout mice display increased serum fasting levels of nonesterified-free FA and show reduced uptake of oleate in isolated adipocytes [11], while cumulative data indicate that impaired CD36 correlates with a large defect in FA uptake by cardiac muscles [12]. These findings suggest that abnormalities in CD36 expression impact susceptibility to certain metabolic diseases such as obesity and insulin resistance [13]. Therefore, an understanding of the mechanism of regulating CD36 expression or CD36 functioning is important for potential therapeutic treatments of metabolic diseases such as obesity and type 2 diabetes [14].
Regulation of CD36-mediated LCFA uptake has been proposed to occur via translocation of CD36 from an intracellular pool to the PM, either in response to insulin or, in muscle cells, to contraction [15, 16]. Subcellular fractionation provided biochemical evidence for insulin-induced redistribution of CD36 from an endocytic to a PM localization in heart and skeletal muscle cells [16, 17]. This redistribution of CD36 strongly resembles that of the well-described insulin-induced glucose transporter 4 (GLUT4) translocation [18, 19].
Chromium (Cr) is an essential nutrient that has been reported to support glucose and lipid metabolism [20]. The Cr complex of picolinic acid (CrPic), which is the most popular dietary supplement form of Cr, has been proposed as a therapeutic agent for treating insulin resistance and diabetes [21]. Investigations into the effects of CrPic on CD36 translocation will therefore be very important for understanding its role in treating diabetes and regulating lipid metabolism. Adipocytes are the primary sites for lipid storage and mobilization. As 3T3-L1 cells differentiate and acquire an adipocyte phenotype, the number of different proteins, which have been implicated in fatty acid transport, among which CD36, increase significantly compared with the values in the undifferentiated preadipocyte state [22]. In this study, we used the 3T3-L1 cell line to examine the effects of insulin and CrPic on CD36 translocation and related influence on LCFA uptake, in comparison with the insulin or CrPic-induced GLUT4 translocation and glucose uptake.
Materials and Methods
Reagents and Cell Lines
3T3-L1 cells (ATCC number CL-173) were cultured using cell culture chemicals purchased from Invitrogen (Breda, the Netherlands). The anti-myc (sc40) and anti-GLUT4 (sc1608) antibodies were obtained from Santa Cruz (Heerhugowaard, the Netherlands). The anti-CD36 antibody JC63.1 and anti-CD36 antibody ab36977 were obtained from Abcam (Cambridge, UK). All fluorescent (donkey antimouse–tetramethyl rhodamine iso-thiocyanate (TRITC); donkey antigoat–TRITC), alkaline phosphatase (AP)-conjugated (goat antirabbit–AP) and horseradish peroxidase (PO)-conjugated secondary antibodies (goat antimouse–PO; donkey antigoat–PO) were purchased from Jackson BioResearch (Amsterdam, the Netherlands), except for the goat antimouse IgA–TRITC antibody (Santa Cruz). [3H]Deoxyglucose ([3H]DOG) and [1-14C]palmitate were obtained from Amersham Biosciences (Roosendaal, the Netherlands). CrPic was purchased from Tokyo Kasei Kogyo (Tokyo, Japan). Insulin, wortmannin, and phloretin were obtained from Sigma (Zwijndrecht, the Netherlands).
Cell Culture and Treatment
The 3T3-L1 cells, of preadipocyte origin, were cultured and propagated in Dulbecco’s modified Eagle’s medium (DMEM) containing glucose (25 mM) and 10% (vol/vol) new born calf serum at 37°C in a 5% CO2 atmosphere. To induce differentiation, 2-day postconfluent 3T3-L1 preadipocytes (designated day 0) were fed with DMEM containing 10% fetal bovine serum (FBS), 1 μg/ml insulin, 1 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine until day 2. Cells were subsequently fed with DMEM supplemented with 10% FBS and 1 μg/ml insulin for 2 days, after which they were fed every other day with DMEM containing 10% FBS. Using this protocol, >80% adipocyte differentiation was achieved. Chinese hamster ovary (CHO) cells stably expressing CD36 (CHO-CD36 cells) and myc-tagged GLUT4 (GLUT4myc; CHO-GLUT4myc cells), which were created by Van Oort et al. [18] were cultured at 37°C and 5% CO2 in F10/Ham’s medium supplemented with 9% FBS, penicillin (100 U/μl), and streptomycin (100 μg/μl).
Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis and Immunoblotting
Total cell lysates were prepared as follows: cells were cultured in 6-well plates for 3 days and washed twice in ice-cold phosphate-buffered saline (PBS) just before lysate preparation. Then cells were scraped in 10 μl ice-cold lysis-buffer (20 mM HEPES, 124 mM NaCl, 4.7 mM CaCl2, 2.5 mM Na2HPO4, 1.2 mM MgSO4, 1 mM EDTA, 0.1 mM benzamidine, 1 μg/ml leupeptin, 1 μg/ml aprotinin) containing 2% Triton-X100, and incubated on ice for 10 min. After centrifugation for 10 min at 13.2 rpm, the protein concentration was determined. Subsequently, equal amounts of protein (30 μg) were incubated at 95°C for 5 min in Laemmli sample buffer and loaded on 7.5% polyacrylamide gels. For GLUT4 detection, Laemmli buffer without β-mercaptoethanol and with 0.025% sodium dodecyl sulfate was used, and samples were not heated prior to loading [18, 21]. After sodium dodecyl sulfate–polyacrylamide gel electrophoresis, proteins were transferred to polyvinylidene fluoride membranes. The membranes were blocked for 1 h in block-buffer [5% bovine serum albumin (BSA) in Tris-buffered saline Tween-20 (TBS-T)] and subsequently incubated with primary antibody against CD36 (ab36977, 1:400), GLUT4 (sc1608, 1:5000), or myc (sc40, 1:5000) in block-buffer for 2 h. Then membranes were washed briefly six times in water and once for 15 min in TBS-T. Secondary antibodies were applied in TBS-T for 1 h (CD36 detection: goat antirabbit–AP 1:5000; myc detection: goat antimouse–PO 1:5000; GLUT4 detection: donkey antigoat–PO 1:5000). Subsequently, membranes were washed six times in water, followed by a 10-min wash in TBS-T. GLUT4myc was visualized by ECL (Amersham Biosciences). For CD36 detection, membranes were washed for 5 min in Tris-sodium-magnesium buffer (TSM: 100 mM Tris HCl, 100 mM NaCl, 10 mM MgAc2, at PH 9.0) and stained by incubation in 20 ml TSM containing 50 μg/μl p-nitro blue tetrazolium chloride (NBT; Boehringer, Mannheim, Germany) and 25 μg/μl 5-bromo-4-chloro-3-indolylphosphatep-toluidine (BCIP; Roche Diagnostics, Almere, the Netherlands). After staining, membranes were washed in methanol and dried. Immunoblots are representative of at least three independent experiments.
Immunofluorescence Microscopy
Cells were grown on 15-mm glass cover slips (Menzel-Glaser, Braunschweig, Germany) in 12-well plates. For some experiments, cells were treated with CrPic, insulin, and/or wortmannin in the following way: cells were washed in serum-free medium once, and then incubated in serum-free medium with or without 10 nM CrPic for 16 h. CrPic was dissolved in dimethyl sulfoxide, which was added to the control samples. The next day, medium was replaced by serum-free medium with or without insulin (100 nM) for 30 min. For phosphatidylinositol-3 kinase (PI3K) inhibition by wortmannin, cells were incubated with wortmannin (200 nM) 30 min prior to insulin incubation, followed by the addition of insulin (100 nM) for 30 min. After brief washing in PBS, cells were fixed at room temperature in 4% paraformaldehyde in PBS for 30 min. After washing twice in PBS, cells were permeabilized for 5 min in PBS containing 0.4% Triton-X100, or washed once more in PBS when permeabilization was not required, and quenched for 10 min in PBS–glycine (50 mM). Subsequently, cells were blocked for 30 min in PBS with 2.5% BSA. Primary antibodies were added in PBS with cold water fish (CWF) gelatin (0.1%; Sigma-Aldrich, St. Louis, MO) and 0.25% BSA for 1 h (JC63.1 1:500; sc1608 and sc40 1:250). Unbound antibodies were removed by washing three times for 5 min in PBS CWF gelatin (0.1%), and secondary antibodies were then added in PBS with CWF gelatin and 0.25% BSA (goat antimouse–TRITC 1:500; donkey antigoat–TRITC 1:250; goat antimouse IgA–TRITC 1:200) for 30 min. Afterward, cells were washed five times in PBS CWF gelatin and mounted on object glasses with mowiol. Samples were analyzed using a Zeiss confocal laser scanning microscope with a C-Apochromat 63×/1.2 Water correction objective. Images are representative of those obtained from at least three independent experiments.
[14C]LCFA and [3H]deoxyglucose Uptake
Cells were cultured on 24-mm glass cover slips in six-well plates and serum-free medium with or without 10 nM CrPic for 16 h. Prior to the substrate uptake experiments, cells were washed twice in PBS. For assessment of the kinetics of cellular LCFA uptake, differentiated 3T3-L1 cells and CHO cells in 1.8 ml PBS were incubated for 10 and 30 min, respectively, with 0.6-ml reaction mixture containing 2.6 μCi [1-14C]palmitate and 0.4 mM unlabelled palmitate complexed to 0.3 mM BSA (with corresponding nonprotein-bound palmitate concentration of 6.9 mM, calculated according to Richieri et al. [23]; preparation of the LCFA/BSA complex was according to Luiken et al. [24]). For assessment of the effects of insulin on cellular LCFA and glucose uptake, cells were incubated with or without wortmannin (200 nM, 30 min prior to insulin incubation) and insulin (100 nM) in 1.8 ml PBS for 30 min at 37°C and 5% CO2. Then 0.6-ml reaction mixture containing the radiolabeled palmitate complexed to BSA and 3.3 μCi [3H]DOG (in a total glucose concentration of 0.4 mM) was added. Subsequently, the cells were incubated at 37°C for 30 min. Uptake was stopped by washing the cells five times in ice-cold stop-buffer (PBS, 0.1% BSA, 0.2 mM phloretin). Cover slips were transferred to clean six-well plates, and cells were lysed by 1-hour incubation in 2 ml scintillation fluid. Scintillation fluid was transferred to vials, and radioactivity was determined using a Packard Tricarb 2300 TR liquid scintillation counter.
Statistical Analysis
The data were expressed as means±SD. The statistical significance of differences between the different groups was determined using Student’s t-test. Values of p < 0.05 were considered statistically significant.
Results
CD36 Expression in 3T3-L1 Cells
To investigate whether CD36 translocation was stimulated by insulin or CrPic in differentiated 3T3-L1 cells (adipocytes), first the expression of endogenous CD36 in these cells, as well as in undifferentiated 3T3-L1 cells (preadipocytes), were examined. CHO cells that were stably transfected with rat CD36 cDNA (CHO-CD36) or GLUT4myc cDNA (CHO-GLUT4myc) were used as positive control cells for GLUT4 or CD36 protein expression and translocation [18]. Since these cells have been reported to be insulin-sensitive and were able to metabolize lipid [25, 26]. GLUT4 and CD36 were expressed in differentiated 3T3-L1 cells, but, as expected, not in undifferentiated 3T3-L1 preadipocytes as detected by immunoblotting (Fig. 1a, b, upper panel).

CD36 and GLUT4 protein expression in 3T3-L1 and CHO cells. Representative immunoblots of cell lysates of undifferentiated 3T3-L1 cells (preadipocytes), differentiated 3T3-L1 cells (adipocytes), and CHO-GLUT4myc cells immunodetected with anti-CD36 (a), anti-GLUT4 (b, upper panel), or anti-myc antibody (b, lower panel). GLUT4myc appeared slightly higher in the gel than endogenous GLUT4 from 3T3-L1 adipocytes because of the myc-tag [18]. Representative images of IF staining of permeabilized 3T3-L1 adipocytes (f, g, h), CHO-CD36 cells (c), or CHO-GLUT4myc cells (d, e), compared with undifferentiated 3T3-L1 preadipocytic cells (i, j, k), using anti-CD36 (JC 63.1) (c, f, i), anti-GLUT4 (d, g, j) and anti-myc (e, h, k) antibody
Subsequently, the cellular localization of CD36, GLUT4, or GLUT4myc in permeabilized 3T3-L1 adipocytes and CHO cells was investigated using immunofluorescence (IF) microscopy. In both cells types, the 3T3-L1 adipocytes and the CHO cells, CD36, and GLUT4 were located intracellularly, but the intracellular distribution pattern of CD36 seemed to be—at least partially—different from that of GLUT4 under permeabilized conditions in both cell types (Fig. 1c–g), an observation that was made recently for the CHO cells [18]. IF microscopy and immunoblotting confirmed that CD36 nor GLUT4 was expressed in undifferentiated 3T3-L1 preadipocytes (Fig. 1a, b, i, j).
Insulin or CrPic-induced Translocation of CD36 or GLUT4 in 3T3-L1 Adipocytes or Transfected CHO Cells
To characterize insulin and CrPic-induced translocation of CD36 or GLUT4 in the 3T3-L1 adipocytes or CHO cells, the PM localization of both GLUT4 and CD36 proteins in the presence or absence of 100 nM insulin or 10 nM CrPic was examined using IF microscopy. To visualize the PM localization of GLUT4, GLUT4myc, or CD36, nonpermeabilized cells were used and treated with antibodies directed against extracellular epitopes of the respective proteins. As for CD36, upon insulin or CrPic stimulation of the 3T3-L1 adipocytes, CD36 translocated from intracellular pools to the PM (Fig. 2b–d) in a similar manner as for GLUT4, for both stimulants (Fig. 2j–l). CrPic showed, however, a less strong effect than insulin in these cells. Also in CHO-CD36 cells, the CrPic-induced increase of CD36 expression to the PM was not as strong as that of insulin (Fig. 2f–h). For comparison, the strong GLUT4 translocations were observed in both the 3T3-L1 adipocytes and CHO cells for both stimulants (Fig. 2j, k, l, n, o, p).

Visualization of insulin- and CrPic-induced CD36 translocation. Representative images of IF staining of cell surface CD36 on nonpermeabilized differentiated 3T3-L1 cells (a–d) and CHO-CD36 cells (e–h), representing control (a, e), insulin-treated (b, f), CrPic-treated (c, g), or CrPic- and insulin-treated (d, h) using anti-CD36 antibody JC63.1. Representative images of IF staining of cell surface GLUT4 and GLUT4myc on non-permeabilized differentiated 3T3-L1 cells (i–l) and CHO-GLUT4myc cells (m–p), representing control (i, m), insulin-treated (j, n), CrPic-treated (k, o), or CrPic- and insulin-treated (l, p) using anti-GLUT4 (i–l) or anti-myc (m–p) antibody
Addition of CrPic together with insulin resulted in a PM localization comparable with the PM localization in insulin-incubated cells of both CD36 and GLUT4 in 3T3-L1 cells and CHO cells.
CrPic (10 or 100 nM) could increase the translocation of CD36 or GLUT4 in 3T3-L1 adipocytes. No difference could be observed for the translocation of CD36 or GLUT4 between the two concentrations. Therefore, in this study, 10 nM was used as the standard concentration for the stimulation of the cells by CrPic (data not shown).
Stimulated Uptake of Radiolabeled LCFA or DOG into 3T3-L1 Adipocytes or Transfected CHO Cells
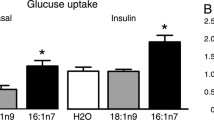
CD36 has been indicated to be a key determinant in cellular LCFA uptake in skeletal and heart muscle [10, 16]. Therefore, whether CD36 expression in nonstimulated cells and after insulin or CrPic-stimulated translocation of CD36 to the PM of 3T3-L1 adipocytes enhanced the cellular uptake of [1-14C]palmitic acid. As expected, the insulin, but not CrPic-stimulated increase in PM localization of GLUT4, that was used as a control, correlated with an approximate threefold increase in 2-[3H]DOG uptake in the 3T3-L1 adipocytes (Fig. 3a) and a relatively 1.2-fold increase of the GLUT4myc transfected CHO cells (Fig. 3b) [18], as compared with nonstimulated cells (Fig. 3a, b). In addition, a small but significant additive effect of CrPic on the insulin-stimulated uptake of radiolabelled glucose in both cells types was observed (Fig. 3a, b), which is in agreement with data obtained by Chen et al. [27]. The uptake of the radiolabelled palmitate in the 3T3-L1 adipocytes was increased approximately 80% upon stimulation with insulin, but no stimulation was observed with CrPic (Fig. 3c). The transfected CHO-CD36 cells did not show any stimulant-induced uptake of [1-14C]palmitic acid (Fig. 3d). An increase in LCFA uptake in CHO-GLUT4myc cells was also not observed upon stimulation with insulin (data not shown) [18]. However, stimulation with CrPic had no effect on LCFA uptake on these cells (data not shown).

Uptake of [3H]DOG or [1-14C]palmitate in 3T3-L1 adipocytes or CHO cells: effects of insulin and/or CrPic. Relative [3H]DOG uptake in differentiated 3T3-L1 adipocytes (a) and CHO-GLUT4myc cells (b) as well as relative radioactive palmitate uptake in differentiated 3T3-L1 adipocytes (c) and CHO-CD36 cells (d), which were control, CrPic-, insulin-, insulin-, and CrPic-treated prior to the addition of [3H]DOG, or [14C]palmitate (complexed to BSA). After cell lysis, the radioactivity was determined and expressed as a multiple relative uptake compared with the uptake in the control group. Results are presented as means of three individual experiments±SD. *#Values of p < 0.05 and ##values of p < 0.01 were considered statistically significant. * indicates a difference between non-CrPic-treated cells and CrPic-treated (10 nM) cells. #indicates a (0 nM CrPic) difference between basal group (0 nM insulin) and insulin-stimulated group (100 nM insulin)
Taken together, insulin-induced GLUT4 or CD36 expression and cell surface localization in the 3T3-L1 adipocytes were sufficient to increase the uptake of glucose and LCFA in these cells. However, as for CrPic, GLUT4 or CD36 translocation is not sufficient to induce an increase of glucose or LCFA uptake in both cell types.
Effects of Wortmannin on Insulin- and CrPic-induced CD36 and GLUT4 Translocation in 3T3-L1 Adipocytes or Transfected CHO Cells
To examine whether the PI3K signal transduction pathway is involved in insulin or CrPic-induced CD36 and GLUT4 translocation in 3T3-L1 cells, the PI3K-inhibitor wortmannin (200 nM) was added to the cells prior to and in the presence of the stimulants. Wortmannin inhibited the insulin-stimulated translocation of GLUT4 in 3T3-L1 adipocytes as well as CHO-GLUT4myc cells completely (Fig. 4j, n). The CD36 translocation stimulated by insulin in CHO-CD36 cells was inhibited by wortmannin, as observed before by Van Oort et al. [18] (Fig. 4f). The translocation of CD36 induced by insulin in the 3T3-L1 adipocytes was inhibited by wortmannin (Fig. 4b), but not as obvious as that for GLUT4 in both cell types (Fig. 4j, n). In comparison, the CrPic-stimulated translocation of CD36 as well as GLUT4 was not inhibited by wortmannin in both cell types (compare Fig. 4a vs. c and b vs. d, e vs. g and f vs, h, i vs. k and j vs. l, m vs. o and n vs. p).

Visualization of insulin and CrPic-induced CD36 translocation in the presence of wortmannin. All cells were treated with wortmannin 30 min prior to incubation with insulin and Cr. a–h Representative images of IF staining of cell surface CD36 on non-permeabilized 3T3-L1 adipocytes (a–d) and CHO-CD36 cells (e–h), which are control (a, e), insulin-treated (b, f), CrPic-treated (c, g), and CrPic- and insulin-treated (d, h) prior to fixation, using anti-CD36 antibody JC63.1. i–p Representative images of IF staining of cell surface GLUT4 and GLUT4myc on nonpermeabilized 3T3-L1 adipocytes (i–l) and CHO-GLUT4myc cells (m–p), which are control (i, m), insulin-treated (j, n), CrPic-treated (k, o), and CrPic- and insulin-treated (l, p) prior to fixation, using anti-GLUT4 (i–l) and anti-myc (m–p) antibody
Effect of Wortmannin on Insulin- or CrPic-induced [14C]LCFA or [3H]DOG Uptake in 3T3-L1 Adipocytes or Transfected CHO Cells
Lastly, the effect of wortmannin on insulin and CrPic-induced LCFA and glucose uptake was investigated. Wortmannin was able to completely inhibit insulin-stimulated glucose uptake in 3T3-L1 adipocytes and CHO cells as well as the LCFA uptake in 3T3-L1 adipocytes (Fig. 5a–c), but not the CrPic’s effects on insulin-stimulated glucose uptake in both cells (Fig. 5a, b).

Uptake of [3H]DOG and [1-14C]palmitate in 3T3-L1 adipocytes and CHO cells in the presence of wortmannin: effects of insulin and/or CrPic. All the cells were treated with wortmannin 30 min prior to insulin incubation. Relative [3H]DOG uptake (a, b) and relative [14C]palmitate uptake (c, d) in 3T3-L1 adipocytes (a, c), CHO-GLUT4myc cells (b) and CHO-CD36 cells (d), which were control, CrPic-treated, insulin-treated, and insulin- and CrPic-treated prior to the addition of [3H]DOG, or [14C]palmitate (complexed to BSA). After cell lysis, the radioactivity was determined and expressed as a multiple relative uptake compared with the uptake in the control group. Results are presented as means of three individual experiments±SD. The statistical significance of differences between the non-CrPic group (0 nM CrPic) and CrPic group (10 nM CrPic) was determined using Student’s t-test. *Values of p < 0.05 were considered statistically significant
Discussion
Recent investigations on skeletal and heart muscle tissues as well as transfected CHO cells have provided evidence for the translocation of CD36 from intracellular pools to the PM in response to either insulin or muscle contraction stimulation [18, 28, 29]. Both under normo- and pathophysiological conditions, adipose tissue plays a crucial and dynamic role not only in fatty acid metabolism but also in glucose metabolism [30]. In adipose cells, the insulin-stimulated glucose uptake is mediated importantly by an enhanced translocation of GLUT4 to the PM [31], a process that is enhanced by the addition of CrPic [27, 32], a dietary supplement that may have a beneficial effect in the treatment of type 2 diabetes. However, the insulin-stimulated uptake of LCFAs by CD36 and the potential beneficial effect herein of CrPic have not been studied yet. In our current studies, we focused on 3T3-L1 adipocytes to study the involvement of translocation of CD36 in LCFA uptake upon insulin- and/or CrPic-stimulation of these cells. In these studies, transfected CHO cells were used in a comparative manner to identify the stimulated translocation process of CD36 and GLUT4, and insulin-stimulated uptake of glucose of the GLUT4-transfected cells.
CD36, as well as GLUT4 protein, is not detected at the PM of 3T3-L1 preadipocytes; however, both proteins are expressed relatively strongly in the PM of 3T3-L1 adipocytes instead. Consequently, in this study, preadipocytes were not analyzed any further. Insulin induced the translocation of CD36 from intracellular pools to PM in both 3T3-L1 adipocytes and CHO-CD36 cells in a similar manner to that of GLUT4. However, in 3T3-L1 adipocytes, but not CHO-CD36 cells, insulin increased the uptake of LCFA. For the CHO-CD36 cells, this result is in accordance with the work by Van Oort et al. [18]. This indicates that in the adipocytes, CD36’s role in the insulin-stimulated uptake of LCFA may resemble that of GLUT4 in the insulin-stimulated uptake of glucose. In the CHO-CD36 cells, however, CD36 expression and PM localization are not sufficient to increase LCFA uptake, whereas insulin-stimulated glucose uptake is driven by GLUT4 in these cells. As proposed, CHO cells may lack a necessary interacting protein or activation step of CD36 to be able to mediate efficient transport of LCFA upon stimulation with insulin [18]. As to CrPic, this stimulant was able to induce the translocation of CD36 to the PM in both 3T3-L1 adipocytes and CHO-CD36 cells, but this translocation did not correlate with an increase in LCFA uptake in either cell type. Therefore, it may be concluded that in 3T3-L1 adipocytes, the mechanism of CrPic-stimulated CD36 translocation is different from that induced of insulin. In addition, it suggests that CrPic acts differently on the translocation of GLUT4 in 3T3-L1 adipocytes, since CrPic alone induces the trafficking to, but not the association of GLUT4 to the PM in these cells [27]. Apparently, in 3T3-L1 adipocytes, CrPic stimulates the incorporation of CD36 into the PM, where it apparently lacks an activation step or interaction partners. In our previous investigation, however, using insulin-resistant 3T3-L1 adipocytes, CrPic was shown to stimulate glucose uptake [33]. Most likely the (patho)physiological status of the cells and culturing conditions are responsible for the difference in CrPic stimulated uptake of glucose, since the insulin-resistant cells were induced to take up glucose by treatment with high glucose (25 mM) and high insulin (100 nM) for 24 h.
As for the signaling pathways involved in the translocation, the PI3K-Akt/PKB pathway was demonstrated to be responsible for the insulin-induced translocation of GLUT4 and CD36 in CHO cells and skeletal and cardiac myocytes [18, 34–36]. Furthermore, the PI3K inhibitor wortmannin abolished insulin-induces translocation of CD36 and GLUT4 as well as the associating increased LCFA and glucose uptake in 3T3-L1 adipocytes. Therefore, the insulin-induced CD36 translocation, as well as the resulting LCFA uptake, appears to be similar to the insulin-induced GLUT4 translocation and glucose uptake in 3T3-L1 adipocytes; both of them are transduced through the PI3K pathway. Subsequently, wortmannin was not able to suppress the effect of CrPic on CD36 translocation in 3T3-L1 adipocytes or CHO cells. At the same time, the CrPic-induced GLUT4 translocation and associated glucose uptake could neither be inhibited by wortmannin. At the same time, the CrPic induced insulin-stimulated GLUT4 translocation and associated glucose uptake could neither be inhibited by wortmannin. This result is coincided with the report showing that CrPic activates the trafficking of GLUT4 to the PM, but does not incorporate the transporter into the PM, in a PI3K-independent manner [27, 37]. Similar effects were reported by Wang et al. [38], who found that Cr increased the insulin receptor kinase activity but did not inhibit protein tyrosine phosphatase (PTP1B) in CHO cells. The signal transduction mechanism by which CrPic increases CD36 translocation in 3T3-L1 adipocytes or CHO cells is unknown. Our previous finding showed that CrPic elevated levels of phosphorylated AMP-activated protein kinase (AMPK) and acetylCoA carboxylase in 3T3-L1 adipocytes [39]. Considering the important role of AMPK in CD36 translocation [4, 15, 28, 36], the AMPK pathway may be involved in the CrPic’s effects on CD36 translocation. This needs further investigation.
In summary, we have demonstrated that insulin-stimulated LCFA uptake correlated with the PM translocation of CD36 in 3T3-L1 cells, most likely, through the involvement of the PI3K pathway. Furthermore, our investigation shows for the first time that CrPic induces translocation of CD36 to the PM, but the translocation does not contribute to an increase in LCFA uptake in both 3T3-L1 adipocytes and CHO cells. The effect of CrPic on CD36 translocation is independent of the PI3K-mediated signal transduction pathway and is thereby different from the insulin-mediated effect on the translocation. In the context of understanding the improved insulin sensitivity of type 2 diabetes patients upon treatment with dietary Cr supplements, further studies are required to elucidate the exact molecular mechanism by which Cr(Pic) induces CD36 translocation to the PM and concomitantly induce LCFA uptake in 3T3-L1 adipocytes.
References
Newsholme EA, Calder P, Yaqoob P (1993) The regulatory, informational, and immunomodulatory roles of fat fuels. Am J Clin Nutr 57:738S–50S
Hamilton JA, Kamp F (1999) How are free fatty acids transported in membranes? Is it by proteins or by free diffusion through the lipids? Diabetes 48:2255–2269
Schaffer JE (2002) Fatty acid transport: the roads taken. Am J Physiol Endocrinol Metab 282:E239–E246
Nickerson JG, Momken I, Benton CR, Lally J, Holloway GP, Han XX, Glatz JF, Chabowski A, Luiken JJ, Bonen A (2007) Protein-mediated fatty acid uptake: regulation by contraction, AMP-activated protein kinase, and endocrine signals. Appl Physiol Nutr Metab 32:865–873
Storch J, Corsico B (2008) The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu Rev Nutr 28:73–95
Chmurzyńska A (2006) The multigene family of fatty acid-binding proteins (FABPs): function, structure and polymorphism. J Appl Genet 47:39–48
Gimeno RE (2007) Fatty acid transport proteins. Curr Opin Lipidol 18:271–276
Doege H, Stahl A (2006) Protein-mediated fatty acid uptake: novel insights from in vivo models. Physiology (Bethesda) 21:259–268
Luiken JJ, Schaap FG, Van Nieuwenhoven FA, Van der Vusse GJ, Bonen A, Glatz JF (1999) Cellular fatty acid transport in heart and skeletal muscle as facilitated by proteins. Lipids 34:S169–S175
Ibrahimi A, Bonen A, Blinn WD, Hajri T, Li X, Zhong K, Cameron R, Abumrad NA (1999) Muscle-specific overexpression of CD36 enhances fatty acid oxidation by contracting muscle, reduces plasma triglycerides and fatty acids, and increases plasma glucose and insulin. J Biol Chem 274:26761–26766
Coburn CT, Knapp FF Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA (2000) Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem 275:32523–32529
Okamoto F, Tanaka T, Sohmiya K, Kawamura K (1998) CD36 abnormality and impaired myocardial long-chain fatty acid uptake in patients with hypertrophic cardiomyopathy. Jpn Circ J 62:499–504
Leprêtre F, Vasseur F, Vaxillaire M, Scherer PE, Ali S, Linton K, Aitman T, Froguel P (2004) A CD36 nonsense mutation associated with insulin resistance and familial type 2 diabetes. Hum Mutat 24:104
Lobo S, Bernlohr DA (2007) Fatty acid transport in adipocytes and the development of insulin resistance. Novartis Found Symp 286:113–121
Luiken JJ, Coort SL, Willems J, Coumans WA, Bonen A, Van der Vusse GJ, Glatz JF (2003) Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes 52:1627–1634
Luiken JJ, Koonen DP, Willems J, Zorzano A, Becker C, Fischer Y, Tandon NN, Van Der Vusse GJ, Bonen A, Glatz JF (2002) Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes 51:3113–3139
Luiken JJ, Dyck DJ, Han XX, Tandon NN, Arumugam Y, Glatz JF, Bonen A (2002) Insulin induces the translocation of the fatty acid transporter FAT/CD36 to the plasma membrane. Am J Physiol Endocrinol Metab 282:E491–E495
Van Oort MM, Van Doorn JM, Bonen A, Glatz JF, Van der Horst DJ, Rodenburg KW, Luiken JJ (2008) Insulin-induced translocation of CD36 to the plasma membrane is reversible and shows similarity to that of GLUT4. Biochim Biophys Acta 1781:61–71
Koonen DP, Glatz JF, Bonen A, Luiken JJ (2005) Long-chain fatty acid uptake and FAT/CD36 translocation in heart and skeletal muscle. Biochim Biophys Acta 1736:163–180
Morris BW, Kouta S, Robinson R, MacNeil S, Heller S (2000) Chromium supplementation improves insulin resistance in patients with type 2 diabetes mellitus. Diabet Med 17:684–685
Wang ZQ, Zhang XH, Russell JC, Hulver M, Cefalu WT (2006) Chromium picolinate enhances skeletal muscle cellular insulin signaling in vivo in obese, insulin-resistant JCR:LA-cp rats. J Nutr 136:415–420
Yang Y, Chen M, Loux TJ, Harmon CM (2007) Regulation of FAT/CD36 mRNA gene expression by long chain fatty acids in the differentiated 3T3-L1 cells. Pediatr Surg Int 23:675–683
Richieri GV, Anel A, Kleinfeld AM (1993) Interactions of long-chain fatty acids and albumin: determination of free fatty acid levels using the fluorescent probe ADIFAB. Biochemistry 32:7574–7580
Luiken JJ, Van Nieuwenhoven FA, America G, Van der Vusse GJ, Glatz JF (1997) Uptake and metabolism of palmitate by isolated cardiac myocytes from adult rats: involvement of sarcolemmal proteins. J Lipid Res 38:745–758
Bogan JS, McKee AE, Lodish HF (2001) Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations. Mol Cell Biol 21:4785–4806
Freeman M, Ekkel Y, Rohrer L, Penman M, Freedman NJ, Chisolm GM, Krieger M (1991) Expression of type I and type II bovine scavenger receptors in Chinese hamster ovary cells: lipid droplet accumulation and nonreciprocal cross competition by acetylated and oxidized low density lipoprotein. Proc Natl Acad Sci USA 88:4931–4935
Chen G, Liu P, Pattar GR, Tackett L, Bhonagiri P, Strawbridge AB, Elmendorf JS (2006) Chromium activates glucose transporter 4 trafficking and enhances insulin-stimulated glucose transport in 3T3-L1 adipocytes via a cholesterol-dependent mechanism. Mol Endocrinol 20:857–870
Chabowski A, Momken I, Coort SL, Calles-Escandon J, Tandon NN, Glatz JF, Luiken JJ, Bonen A (2006) Prolonged AMPK activation increases the expression of fatty acid transporters in cardiac myocytes and perfused hearts. Mol Cell Biochem 288:201–212
Bonen A, Luiken JJ, Arumugam Y, Glatz JF, Tandon NN (2000) Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem 275:14501–14508
Kuniyasu A, Hayashi S, Nakayama H (2002) Adipocytes recognize and degrade oxidized low density lipoprotein through CD36. Biochem Biophys Res Commun 295:319–323
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806
Brautigan DL, Kruszewski A, Wang H (2006) Chromium and vanadate combination increases insulin-induced glucose uptake by 3T3-L1 adipocytes. Biochem Biophys Res Commun 347:769–773
Wang YQ, Yao MH (2009) Effects of chromium picolinate on glucose uptake in insulin-resistant 3T3-L1 adipocytes involve activation of p38 MAPK. J Nutr Biochem 20:982–991
Luiken JJ, Coort SL, Koonen DP, Van der Horst DJ, Bonen A, Zorzano A, Glatz JF (2004) Regulation of cardiac long-chain fatty acid and glucose uptake by translocation of substrate transporters. Pflugers Arch 448:1–15
Rauch C, Loughna P (2005) C2C12 skeletal muscle cells exposure to phosphatidylcholine triggers IGF-1 like-responses. Cell Physiol Biochem 15:211–224
Habets DD, Coumans WA, Voshol PJ, den Boer MA, Febbraio M, Bonen A, Glatz JF, Luiken JJ (2007) AMPK-mediated increase in myocardial long-chain fatty acid uptake critically depends on sarcolemmal CD36. Biochem Biophys Res Commun 355:204–210
Pattar GR, Tackett L, Liu P, Elmendorf JS (2006) Chromium picolinate positively influences the glucose transporter system via affecting cholesterol homeostasis in adipocytes cultured under hyperglycemic diabetic conditions. Mutat Res 610:93–100
Wang H, Kruszewski A, Brautigan DL (2005) Cellular chromium enhances activation of insulin receptor kinase. Biochemistry 44:8167–8175
Wang YQ, Dong Y, Yao MH (2009) Chromium picolinate inhibits resistin secretion in insulin-resistant 3T3-L1 adipocytes via activation of amp-activated protein kinase. Clin Exp Pharmacol Physiol 36:843–849
Acknowledgments
This study was supported financially by a Short Stay (3 months) Fellowship for Ph.D. students from China, awarded by Utrecht University (Utrecht, the Netherlands), grants-in-aid for Shanghai Leading Academic Discipline Project (B119), the Shanghai Committee of Science and Technology (10ZR1402100, 10XD1400400), China National Science and Technology Major Project for Drug Discovery (2009ZX09303-006).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Y., Van Oort, M.M., Yao, M. et al. Insulin and Chromium Picolinate Induce Translocation of CD36 to the Plasma Membrane Through Different Signaling Pathways in 3T3-L1 Adipocytes, and with a Differential Functionality of the CD36. Biol Trace Elem Res 142, 735–747 (2011). https://doi.org/10.1007/s12011-010-8809-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-010-8809-8