
Abstract
Xylo-oligosaccharides with high value could be obtained by acidolysis of lignocellulosic biomass with acetic acid, which was an urgent problem to solve for the separation of acetic acid from crude xylo-oligosaccharides solution. Four neutralizers, CaCO3, CaO, Na2CO3, and NaOH, were used for in situ chemically locking the acetic acid in the acidolyzed hydrolysate of corncob. The chemically locked hydrolysate was analyzed and compared using vacuum evaporation and spray drying. After CaCO3, CaO, Na2CO3, and NaOH treatment, the locking rates of acetic acid were 92.62%, 94.89%, 95.05%, and 95.58%, respectively, and 39.55 g, 41.13 g, 41.78 g, and 41.87 g of the compound of xylo-oligosaccharide and acetate were obtained. Sodium neutralizer had lesser effect on xylo-oligosaccharide content, and Na2CO3 was the best chemical for locking acetic acid among these four neutralizers. This process provides a novel method for effectively utilizing acetic acid during the industrial production of xylo-oligosaccharides via acetic acid.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lignocellulosic biomass from agricultural and forestry wastes represent huge renewable resources, and the conversion of these renewable resources into commercially valuable products such as bio-based fuels, materials, and chemicals has been the focus of lignocellulosic biomass biorefineries [1,2,3]. Hemicellulose is a non-linear polysaccharide composed of pentose and hexose, including xylose, arabinose, galactose, and glucose. Owing to its highly branched and amorphous nature, hemicellulose is non-crystalline and therefore more easily hydrolyzed than cellulose [4, 5]. The hemicellulose of gramineous plants possesses a xylan backbone [6]; hence, pretreatment of lignocellulosic biomass can result in effective degradation of hemicellulose xylan to xylo-oligosaccharides (XOS) [7]. XOS is more beneficial for stimulating the growth and proliferation of intestinal probiotics such as Bifidobacterium than other oligosaccharides such as fructo-oligosaccharides [8]. Therefore, XOS is considered a soluble dietary fiber with prebiotic activity, which assists in improving intestinal function, enhances immune function, and possesses antibacterial and other health benefits [9, 10]. The market price of XOS was about 22–50 USD/kg; however, the specific price depends on the actual purity of the XOS products [11].
In general, strong inorganic acids can effectively catalyze and cleavage the glycosidic bonds between adjacent xylose units in the xylan skeleton, but their action is random rather than selective. Therefore, the major disadvantage of this process is the large production of monosaccharides sugars and undesirable by-products [12, 13]. Compared with the use of strong inorganic acid for acidolysis, Zhang et al. used acetic acid (AA) for the acidolysis of viscose fiber processing waste xylan and got a smaller amount of xylose and furfural to prove a somewhat higher selectivity of AA catalysis [14]. Besides, they also showed that compared with inorganic acids with the same pH value, acidolysis of xylan with AA can obtain a higher yield of XOS, which is more suitable for preparing XOS from corncob hemicellulose [15]. Moreover, AA is an attractive pretreatment reagent for lignocellulosic biomass because of its easy availability and volatility, and can be recycled from the pretreatment solution via vacuum evaporation and electrodialysis [16].
Excessive degradation and heterogeneity of the hemicellulose structure during the preparation of XOS from lignocellulosic biomass acidolyzed with AA inevitably leads to the formation of inhibitory by-products such as AA, furfural, and hydroxymethylfurfural (HMF) in the AA hydrolysate [15, 17], which are toxic to animals; Niu et al. used AA to induce ulcerative colitis in mice, as it injured the colon mucosa [18], and Gill et al. observed that furan compounds were carcinogenic for rodent liver [19]. Therefore, these compounds in the AA hydrolysate of lignocellulosic biomass might affect the application of XOS products in medicine and health care. Hence, an effective technique for effectively removing the inhibitors from the AA hydrolysate of lignocellulosic biomass is urgently required. Gurram et al. observed that detoxification of dilute acid-pretreated ponderosa pine slurry via sequential polyelectrolyte and resin-wafer electrodeionization yielded promising results, and the removal rates of AA, furfural, and 5-HMF reached 77%, 74%, and 60%, respectively [20]. Jeong et al. also showed that furfural and 5-HMF in wood hydrolysate can be removed using charcoal [21]. Ke-KeCheng et al. detoxified the acid hydrolysate of bagasse via electrodialysis and removed 90% AA from the hydrolysate [22]. Thus, the process and technology of removing the inhibitors from the hydrolysate of lignocellulosic biomass are well developed.
Although the acidic hydrolysate of lignocellulosic biomass contains a variety of degraded compounds, AA was certainly the biggest inhibitor because of at least 5% exogenously AA catalyst loading besides the newly generated portion during acidic hydrolysis of hemicellulose constitute from corncob. Currently, the commonly used AA removal methods include liquid extraction, direct distillation, surfactant membrane extraction, anion exchange, electrodialysis, precipitation, and adsorption [23]. For a long time, most processes have aimed to remove and recover AA from hydrolysate in the form of liquid or steam. For example, Aymn et al. successfully extracted AA from green liquor pre-pulping hardwood extract using the liquid-liquid extraction method, and completed the removal of AA after acidolysis and before fermentation [24]; Huang et al. recovered 76% AA from the AA hydrolysate of poplar via vacuum evaporation [25]. Removal of AA by precipitation is controversial, as it resulted in the production of large amounts of solid waste, and the use of precipitants considerably increased the cost of the process [23]. Therefore, chemicals with added value can be obtained while removing AA via the precipitation method, which can avoid the generation of solid waste.
Traditionally, calcium salts react with acids to form precipitates during deacidification. As acetate is mostly soluble in water, precipitation of acetates is difficult. However, acetate can be harvested by spray drying. Acetates such as calcium acetate and sodium acetate are good feed stabilizers, which can be widely added to animal feed along with XOS. Based on the precipitation method, calcium salt or sodium salt was added to AA hydrolysate of lignocellulosic biomass to convert AA in crude XOS liquid into available acetate for the feed to “lock” the AA, which can effectively save the energy required for removing AA via distillation and electrodialysis. At the same time, the compound of xylo-oligosaccharides and acetate (CXA) can be formed, which effectively solves the problems associated with AA as a by-product.
The purpose of this study was to control the presence of AA in the AA hydrolysate. Toward this, a process of chemically locking AA in the AA hydrolysate of corncob (AHC) using calcium salt and sodium salt neutralizers, leading to the formation of CXA, was proposed. The effects of chemical locking of the neutralizer on XOS, AA, furfural, and HMF in AHC were studied. CXA was prepared by spray drying. In general, the use of neutralizers to chemically lock AA in AHC provides a promising reference for the industrial separation of AA while preparing XOS with AA acidolysis.
Materials and Methods
Materials
The crude XOS solution was derived from the AHC. Corncob was collected from Jiangsu Kangwei Biologic Co., Ltd. and contained 32.86% glucan, 31.53% xylan, 4.68% arabinose, 4.42% acid-soluble lignin, and 19.03% acid-insoluble lignin. AA was purchased from Nanjing Chemical Co., Ltd. (Jiangsu province, China). CaCO3, CaO, Na2CO3, NaOH, (CH3COO)2Ca, and CH3COONa were purchased from Sinopharm Chemical Reagent Co., Ltd. (China). All chemicals used in the experiment were of analytical grade.
AA Acidolysis of Corncob
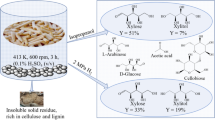
Corncob was acidolyzed using 5% (v/v) and 20% (v/v) AA, respectively. Zhang et al. prepared XOS by pretreating corncob with AA [15]. Based on this and to ensure the yield of XOS, the reaction temperature was set to 150 °C and the AA concentration to 20% (V/V) for 30 min. When the concentration of AA was 5% (v/v), the holding time was extended to 65 min. Corncob was mixed with AA at a solid-liquid ratio of 1:10, and 5% (v/v) AA acidolysis was performed in a 30-mL stainless steel reaction tube, while 20% (v/v) AA acidolysis was performed in a 1.25-L high-temperature reaction kettle. The temperature of the acidolysis process was controlled using a constant temperature oil bath. After acidolysis, the reaction kettle was put in cold water for rapid cooling, following which the AA acidolysis products of corncob were collected. After centrifugation, AHC containing XOS was obtained. The concentrations of XOS (X2-X6), AA, furfural, and HMF in the 5% (v/v) AHC were 14.39 g/L, 52.49 g/L, 0.80 g/L, and 0.22 g/L, respectively, while the same in the 20% (v/v) AHC of corncob were 17.55 g/L, 209.52 g/L, 1.20 g/L, and 0.41 g/L, respectively.
Chemical Locking of AA in AHC
The chemical locking of AA was performed in a water bath at 60 °C. Based on the amount of AA in 5% (v/v) AHC, the amounts of four chemical locking agents, CaCO3, CaO, Na2CO3, and NaOH, to be added were calculated; the final addition amount was based on adjusting the pH of the hydrolysate to neutrality (pH was about 7). Then, the original AHC and the hydrolysate with locking agents were rotated and evaporated at 60 °C and under negative pressure (10 ± 1.0 KPa).
Spray-Drying of AHC
The 20% (v/v) AHC neutralized with CaCO3, CaO, Na2CO3, and NaOH was spray-dried to obtain the finished product powder of CXA, which was used to verify the chemical locking effect of the four neutralizers on the AA in the AHC. The spray-drying air inlet temperature was 170 °C and the outlet air temperature was 70–80 °C.
Analytical Methods
The solid content in AA hydrolysate was determined using an infrared moisture analyzer. Acetate, furfural, and HMF in AHC were detected using high-performance liquid chromatography (HPLC, Agilent 1260), and analyzed using an Aminex Bio-Rad HPX-87H column and a reflective index detector. The mobile phase was 5 mmol/L H2SO4 with a flow rate of 0.6 mL/min. High-performance anion-exchange chromatography, coupled with pulsed amperometric detection (HPAEC-PAD) (Dionex ICS-3000), was used to determine the XOS (including xylobiose (X2), xylotriose (X3), xylotetraose (X4), xylopentaose (X5), and xylohexaose (X6)) levels in the AA hydrolysate. CarboPacTM PA200 was the analytical column used. The mobile phase consisted of 100 mmol/L NaOH and 500 mmol/L NaOAc at the flow rate of 0.3 mL/min [14, 26].
The concentration multiple of acetate and HMF, residual rate of acetate and furfural during vacuum evaporation, locking rate of AA, and total recovery rate of CXA after spray drying were calculated as follows: (1–4)
CA/H in Eq. (1) represents the concentration of acetate or HMF in the various AHC corresponding to the change in concentration factor during vacuum evaporation; Ci represents the initial concentration of acetate or HMF in AHC after adding different neutralizers.
CA/F in Eq. (2) represents the concentration of acetate or furfural in the various AHC corresponding to the change in concentration factor during vacuum evaporation; Vv represents the volume of the remaining AHC corresponding to the concentration factor; Vi is the initial volume of AHC after adding different neutralizers.
Results and Discussion
Comparison of the Chemical Locking Effects of Four Neutralizers on AA in Low-Concentration AHC
Four neutralizers, CaCO3, CaO, Na2CO3, and NaOH, were used to chemically lock AA in 5% AHC. The four neutralizers combined with AA to form calcium acetate or sodium acetate, and the chemical reaction is shown as Eqs. (5)–(8). This indicated that AA was locked in the form of acetate in AHC, which is an excellent stabilizer for feed. Miller et al. has demonstrated that sodium acetate has negligible toxic effect on the growth and milk yield of dairy cows [27]. Therefore, forming CXA by converting AA in AHC into acetate will be easy, which will no longer exert toxic effect on animal growth. AA is a volatile chemical. Usually, part of the AA in hydrolysates can be distilled off via vacuum evaporation. Therefore, the vacuum evaporation method was used to verify the chemical locking effect of CaCO3, CaO, Na2CO3, and NaOH on AA in AHC. As shown in Fig. 1a and b, after the original AHC and the chemically locked hydrolysate were evaporated under reduced pressure, the acetate concentration in the original AHC increased slowly with the concentration factor. Correspondingly, the residual rate of AA in the original AHC gradually decreased during the vacuum evaporation process. Within the concentration factor of 5, the residual rate of AA was only 36.95%, which indicated that most of the AA in the original AHC was removed via distillation during evaporation under reduced pressure; however, separation from the hydrolysate was difficult, as the boiling point of AA was higher than that of water. Therefore, distillation is not a feasible approach for removing AA [24, 28]. However, the concentration of AA in the hydrolysate chemically locked by the four neutralizers increased with the concentration factor, and the concentration multiples of AA in the hydrolysate corresponding to CaCO3, CaO, Na2CO3, and NaOH increased approximately synchronously with the concentration factor. With the increase in concentration multiple, the residual rate of acetate in various AHCs supplemented with the neutralizer also decreased, which may be due to the hydrolysis of sodium acetate or calcium acetate formed after chemical locking, producing AA during vacuum evaporation, thereby gradually reducing acetate concentration [29]. However, this was like the changing trend of pure sodium acetate and calcium acetate solution in Fig. 1c during vacuum evaporation. In other words, the AA in the AHC was locked as acetate after adding the neutralizer; hence, it was concentrated with loss of water due to vacuum evaporation, indicating that the chemical locking of neutralizer was successful. Obviously, after the chemical locking of the four neutralizers, the concentration of AA in the hydrolysate increased with the concentration multiple, showing the characteristics of the exponential function. Among these, the concentration of acetate in AHC after chemical locking by CaCO3 and CaO showed a trend of increasing and then decreasing, while the concentration of acetate in AHC after chemical locking by Na2CO3 and NaOH showed a more obvious upward trend. We compared the changes in acetate concentration and residual rate during vacuum evaporation between acetate formed after chemical locking and pure acetate (Fig. 1a–c). We believe that neutralizer can be used for chemical locking of AA in AHC, and sodium salt was more advantageous and stable for the chemical locking of AA.

Comparison of the effects of four neutralizers on AA in AHC. a Changes in acetate concentration in the AHC; b The residual rate of acetate during vacuum evaporation; c Concentration changes of pure sodium acetate and calcium acetate during vacuum evaporation
Effects of Four Neutralizers on Inhibitors and XOS in Low-Concentration AHC
The concentrations of furfural and HMF in the AHC were also monitored during evaporation under reduced pressure. The variation in furfural concentration is shown in Fig. 2a, and the initial amount of furfural in AHC was 0.8 g/L. However, after chemical locking of CaCO3, CaO, Na2CO3, and NaOH, the content of furfural in AHC decreased significantly to 0.36 g/L, 0.22 g/L, 0.27 g/L, and 0.25 g/L respectively. Compared to that in the original AHC, the furfural content decreased by 55.00%, 72.50%, 66.25%, and 68.75% respectively, which was due to the adsorption of some toxins in the hydrolysate by the added salts such as CaCO3 during the chemical locking process [30], thereby reducing the furfural concentration. The change in the residue rate of furfural in the AHC during vacuum evaporation is shown in Fig. 2b. The furfural in the original AHC was completely removed when the concentration factor was 2.14, but after chemical locking by CaCO3, CaO, Na2CO3, and NaOH, furfural residue in AHC disappeared at concentration factor was less than 1.42. In general, addition of neutralizer to AHC to chemically lock AA promoted the removal of furfural from the hydrolysate.

Evaporation kinetics of furfural and HMF in AHC during vacuum evaporation. a Change in furfural concentration in the AHC; b The residue rate of furfural; c Change in HMF concentration in the AHC; d The concentration multiple of HMF
The change in HMF during vacuum evaporation is shown in Fig. 2c. Like furfural, the content of HMF in AHC after addition of neutralizer was lesser than that in original AHC, and CaCO3, CaO, Na2CO3, and NaOH reduced HMF by 26.61%, 63.58%, 61.60%, and 55.40% respectively. Fatehi et al. observed that precipitated calcium carbonate can adsorb furfural in the hydrolysate, and the adsorption capacity of 50 mg/g [31]. Therefore, we reasonably speculated that the calcium salt and sodium salt added to the AHC adsorbed furan compounds, and that the neutralizer could not only lock the AA, but also exert obvious detoxification effects on inhibitors in the hydrolysate. Furthermore, as shown in Fig. 2d, with increase in concentration factor, the residual amount of HMF in the original AHC increases rapidly due to its low volatility. However, the amount of HMF in the hydrolysate showed a slower upward trend after the addition of the four neutralizers than the original AHC. In other words, neutralizer addition exerts a significant inhibitory effect on HMF, indicating that this method can effectively control the inhibitor content in the hydrolysate and provides favorable conditions for the subsequent purification of XOS.
XOS was one of the final products obtained after the pretreatment of corncob with AA. The effect of neutralizer on the XOS content in the hydrolysate has attracted considerable attention. As shown in Fig. 3, after the AA in the hydrolysate was chemically locked by CaCO3, CaO, Na2CO3, and NaOH, the distribution of each component was not affected. The components of XOS were mainly xylobiose, xylotriose, and xylotetraose, while the proportions of xylopentaose and xylohexaose were relatively small. The addition of the four neutralizers had little effect on the content of XOS in AHC. However, calcium salt has extremely strong adsorption due to the strong positive charge of calcium ions as a widely used adsorbent [32]. Fatehi et al. used porous precipitated calcium carbonate to adsorb the pre-hydrolysates from pulp mills and observed that precipitated calcium carbonate adsorbs XOS, lignin, furfural, and other compounds in the pulp pre-hydrolysate [31]. This proved that the CaCO3 had an adsorption capacity for polysaccharides, so CaCO3 has an adsorption effect on the XOS in AHC resulted in a slight decrease of corresponding XOS content. Similarly, CaO has also slightly reduced the XOS content in AHC due to adsorption. However, Na2CO3 and NaOH had almost no adsorption effect on XOS, which resulted in minimal change in the content of XOS in the AHC with the addition of Na2CO3 and NaOH, and thus exhibited a higher XOS content than AHC added with CaCO3 and CaO. In addition to adsorption, the error of instrument detection was also one of the factors that affected the difference in XOS content. The fluctuation of XOS content in AHC caused by the addition of neutralizers is shown in Fig. 3, and these fluctuations remained within the acceptable error range and could be desorbed by ultrasonication and other means [33].

Effect of neutralizer on XOS concentration in AHC
In a nutshell, addition of neutralizers to AHC to realize the chemical locking of AA was feasible. The process achieved chemical locking of AA and inhibition of furan compounds without affecting the total amount of XOS, thereby achieving effective purification of crude XOS solution.
Effect of Locking AA with Neutralizer on the Preparation of XOS Crude Products Using Subsequent Spray-Drying
To verify the role of the chemical locking effect of the neutralizers on CXA formation, 20% AHC chemically locked by the neutralizers was used for spray-drying to obtain the CXA. As shown in Fig. 4, after adding the four neutralizers to 20% AHC, the changes in XOS, acetate, furfural, and HMF levels in 20% AHC were like those in 5% AHC. Upon addition of the four neutralizers, the AA in AHC was chemically locked into calcium acetate or sodium acetate. Acetate is a strong electrolyte and can be completely ionized in water, while AA is a weak acid and cannot be completely ionized in water. Therefore, the concentration of acetate in each neutralized hydrolysate obtained using HPLC was higher than that in the original AHC. This also proved that the addition of the four neutralizers resulted in chemical locking of AA in the original AHC. Among the four neutralizers, the chemical locking effect of the sodium salt on AA was better than that of the calcium salt in terms of changes in the concentration of acetate and XOS, while the detoxification effect of calcium salt on furan compounds was more obvious, which was attributed to the strong adsorption capacity of calcium salt. Regarding the effect on XOS content, we believe that sodium salt exerts the least adverse effect during chemical locking of AA in AHC.

Chemical composition in the 20% AHC before spray-drying
Spray-drying of the hydrolysate clearly showed the chemical locking effect of the four neutralizers on AA in AHC. As shown in Fig. 5a, after chemical locking by CaCO3, CaO, Na2CO3, and NaOH, the XOS level in 20% AHC was 4.48%, 4.52%, 5.03%, and 4.49%, while the acetate content was 54.72%, 59.11%, 59.54%, and 56.16%, respectively; the furfural content was 0.01–0.10%, and the HMF content was 0.03–0.22% (calculated based on the solid content in the hydrolysate). The components of the dry powder after spray-drying are shown in Fig. 5b. The chemical locking of CaCO3, CaO, Na2CO3, and NaOH corresponded to 4.38%, 4.31%, 4.86%, and 4.45% XOS and 53.25%, 56.04%, 57.93%, and 55.23% acetate, respectively (calculated based on the dry powder quality). However, the content of acetate in the dry powder obtained after spray-drying the original AHC was only 10.89%. In addition, furfural and HMF volatilize at high temperature during spray-drying, because of which furfural and HMF were not present in all dry powders. Based on the purity of XOS and acetate, the ratio of XOS and acetate in the materials before and after spray-drying was similar, which indicated that less XOS and salt were lost during spray-drying. More importantly, spray-drying proved that AA in AHC was chemically locked by the four neutralizers to produce acetate.

Content of components in the material before and after spray-drying. a Components of AHC before spray-drying; b Components of the powder obtained after spray-drying
Mass balance was performed after spray-drying. The evaporation and spray-drying experiments of each batch of XOS showed absence of decomposition and coking changes during the above-mentioned concentration and pulverization processes. After spray-drying, complete collection of the dry powder resulted in total recovery rate of 95–98%. Therefore, the experiments involving the four neutralizers were based on the 95% dry powder recovery rate. As shown in Fig. 6, 35.45 g CaCO3, 24.78 g Cao, 35.43 g Na2CO3, and 26.90 g NaOH were added to four vessels containing 200 mL 20% AHC, respectively, and the XOS content in the hydrolysate after chemical locking was 3.20 g, 3.21 g, 3.49 g, and 3.28 g, while the acetate content in the hydrolysates was 39.46 g, 40.25 g, 40.55 g, and 40.50 g, respectively. After spray-drying, 39.55 g, 41.13 g, 41.78 g, and 41.87 g CXA was harvested from the hydrolysates treated with CaCO3, CaO, Na2CO3, and NaOH, respectively, as a dry powder. The acetate recovery rates were 92.62%, 94.89%, 95.05%, and 95.58%, respectively, while the content of acetate in the dry powder obtained after spray-drying the original AHC was only 5.49%, which strongly indicated that the addition of the four neutralizers exerted good locking effect on AA in the hydrolysate, and the recovery rate of acetate in the dry powder represents the locking rate of AA. In addition, based on the results of previous concentration experiments and spray-drying, the chemical locking effect of Na2CO3 was found to be the best among the four neutralizers, as it showed higher acetate locking rate and stable yield of CXA. However, NaOH is a strong alkali, which renders it unsuitable for large-scale industrial applications in animal feed field owing to safety issues. Therefore, Na2CO3 was considered as the most suitable chemical for the chemical locking of the AA in AHC among the four neutralizers, which could effectively remove and utilize the AA in the AA hydrolysate of lignocellulosic biomass, controlling the safety of XOS products obtained by acidolysis of lignocellulosic biomass with AA.

Mass balance
Conclusions
In conclusion, a method for chemically locking AA was implemented by adding neutralizer to the AHC and obtaining CXA that can be used as prebiotic product. The chemical locking effects of CaCO3, CaO, Na2CO3, and NaOH on AA in AHC were studied. After comparing the effects of neutralizers on AA, XOS, and inhibitors in the AHC, Na2CO3 appeared to be the best for chemically locking AA in the AHC, with AA locking rate of 95.05%. This study provides a practical reference for the industrial preparation, separation, and purification of XOS products from lignocellulosic biomass by AA acidolysis.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Fitzpatrick, M., Champagne, P., Cunningham, M. F., & Whitney, R. A. (2010). A biorefinery processing perspective: treatment of lignocellulosic materials for the production of value-added products. Bioresource Technology, 101(23), 8915–8922.
Stoklosa, R. J., Orjuela, A. D. P., Sousa, L. D. C., Uppugundla, N., Williams, D. L., Dale, B. E., Hodge, D. B., & Balan, V. (2017). Techno-economic comparison of centralized versus decentralized biorefineries for two alkaline pretreatment processes. Bioresource Technology, 226, 9–17.
Guo, Z., Zhang, L., Zhang, L., Yang, G., & Xu, F. (2018). Enhanced enzymatic hydrolysis by adding long-chain fatty alcohols using film as a structure model. Bioresource Technology, 249, 82–88.
Jönsson, L. J., & Martín, C. (2015). Pretreatment of lignocellulose: formation of inhibitory by-products and strategies for minimizing their effects. Bioresource Technology, 199, 103–112.
Otieno, D. O., & Ahring, B. K. (2012). The potential for oligosaccharide production from the hemicellulose fraction of biomasses through pretreatment processes: xylooligosaccharides (XOS), arabinooligosaccharides (AOS), and mannooligosaccharides (MOS). Carbohydrate Research, 360, 84–92.
Peng, F., Ren, J. L., Xu, F., & Sun, R. C. (2011). Sustainable Production of Fuels, Chemicals, and Fibers from Forest Biomass. Chemicals from Hemicelluloses: A Review, 1067, 219–259.
Zhang, X., Zhang, W., Lei, F., Yang, S., & Jiang, J. (2020). Coproduction of xylooligosaccharides and fermentable sugars from sugarcane bagasse by seawater hydrothermal pretreatment. Bioresource Technology, 309, 123385.
Mäkeläinen, H., Forssten, S., Saarinen, M., Stowell, J., & Ouwehand, A. C. (2010). Xylo-oligosaccharides enhance the growth of bifidobacteria and Bifidobacterium lactis in a simulated colon model. Beneficial Microbes, 1(1), 81–91.
Carvalho, A. F. A., Neto, P. D. O., Silva, D. F. D., & Pastore, G. M. (2013). Xylo-oligosaccharides from lignocellulosic materials: Chemical structure, health benefits and production by chemical and enzymatic hydrolysis. Food Research International, 51(1), 75–85.
Li, Z., Summanen, P. H., Komoriya, T., & Finegold, S. M. (2015). In vitro study of the prebiotic xylooligosaccharide (XOS) on the growth of Bifidobacterium spp and Lactobacillus spp. International Journal of Food Sciences and Nutrition, 66(8), 919–922.
Chen, M. H., Bowman, M. J., Dien, B. S., Rausch, K. D., Tumbleson, M. E., & Singh, V. (2014). Autohydrolysis of Miscanthus x giganteus for the production of xylooligosaccharides (XOS): kinetics, characterization and recovery. Bioresource Technology, 155, 359–365.
Sun, H. J., Yoshida, S., Park, N. H., & Kusakabe, I. (2002). Preparation of (1→4)-β- d -xylooligosaccharides from an acid hydrolysate of cotton-seed xylan: suitability of cotton-seed xylan as a starting material for the preparation of (1→4)-β- d -xylooligosaccharides. Carbohydrate Research, 337(7), 657–661.
Akpinar, O., Erdogan, K., Bakir, U., & Yilmaz, L. (2010). Comparison of acid and enzymatic hydrolysis of tobacco stalk xylan for preparation of xylooligosaccharides. LWT-Food Science And Technology, 43(1), 119–125.
Zhang, H., Zhou, X., Xu, Y., & Yu, S. (2016). Production of xylooligosaccharides from waste xylan, obtained from viscose fiber processing, by selective hydrolysis using concentrated acetic acid. Journal of Wood Chemistry and Technology, 37, 1–9.
Zhang, H., Xu, Y., & Yu, S. (2017). Co-production of functional xylooligosaccharides and fermentable sugars from corncob with effective acetic acid prehydrolysis. Bioresource Technology, 234, 343–349.
Yu, L., Guo, Q., Hao, J., & Jiang, W. (2000). Recovery of acetic acid from dilute wastewater by means of bipolar membrane electrodialysis. Desalination, 129(3), 283–288.
Li, H., Kim, N. J., Jiang, M., Kang, J. W., & Chang, H. N. (2009). Simultaneous saccharification and fermentation of lignocellulosic residues pretreated with phosphoric acid-acetone for bioethanol production. Bioresource Technology, 99, 3245–3251.
Niu, X., Fan, T., Li, W., Huang, H., Zhang, Y., & Xing, W. (2013). Protective effec t of sanguinarine against acetic acid-induced ulcerative colitis in mice. Toxicology and Applied Pharmacology, 267(3), 256–265.
Gill, S., Bondy, G., Lefebvre, D. E., Becalski, A., Kavanagh, M., Hou, Y., Turcotte, A. M., Barker, M., Weld, M., & Vavasour, E. (2010). Subchronic oral toxicity study of furan in Fischer-344 rats. Toxicologic Pathology, 38(4), 619–630.
Gurram, R. N., Datta, S., Lin, Y. J., Snyder, S. W., & Menkhaus, T. J. (2011). Removal of enzymatic and fermentation inhibitory compounds from biomass slurries for enhanced biorefinery process efficiencies. Bioresource Technology, 102(17), 7850–7859.
Jeong, H., Yong, S. K., Lee, J., Chea, K. S., & Lee, S. M. (2016). Removal of 5-hydroxymethylfurfural and furfural in sugar hydrolysate by wood charcoal treatment. Journal of the Korean Wood Science & Technology, 2016(44), 705–715.
Cheng, K. K., Cai, B. Y., Zhang, J. A., Ling, H. Z., Zhou, Y. J., Ge, J. P., & Xu, J. M. (2008). Sugarcane bagasse hemicellulose hydrolysate for ethanol production by acid recovery process. Biochemical Engineering Journal, 38(1), 105–109.
Lei, Z., Li, C., Li, Y., & Chen, B. (2004). Separation of acetic acid and water by complex extractive distillation. Separation and Purification Technology, 36(2), 131–138.
Aymn, A., Peter, V. W. G., & Byung-Hwan, U. (2018). Acetic acid removal from pre-pulping wood extract with recovery and recycling of extraction solvents. Applied Biochemistry and Biotechnology, 187, 378–395.
Huang, K., Luo, J., Wu, Y., & Xu, Y. (2019). β-Factor based separation characteristics of bio-derived chemicals present in lignocellulosic hydrolysates using vacuum distillation. ACS Sustainable Chemistry & Engineering, 7(2), 2406–2413.
Zhou, X., Zhao, J., Zhang, X., & Xu, Y. (2019). An eco-friendly biorefinery strategy for xylooligosaccharides production from sugarcane bagasse using cellulosic derived gluconic acid as efficient catalyst. Bioresource Technology, 289, 121755.
Miller, W. J., & Allen, N. N. (1955). The effect of sodium acetate feeding on milk and fat yield, blood sugar, and blood ketones of dairy cows. Journal of Dairy Science, 38(3), 310–312.
Kim, S. J., Kwon, H. S., Kim, G. H., & Um, B. H. (2015). Green liquor extraction of hemicellulosic fractions and subsequent organic acid recovery from the extracts using liquid–liquid extraction. Industrial Crops and Products, 67, 395–402.
Fournier, P., Oelkers, E. H., Gout, R., & Pokrovski, G. (1998). Experimental determination of aqueous sodium-acetate dissociation constants at temperatures from 20 to 240°C. Chemical Geology, 151(1-4), 69–84.
Fatehi, P., Hamdan, F. C., & Ni, Y. (2013). Adsorption of lignocelluloses of pre-hydrolysis liquor on calcium carbonate to induce functional filler. Carbohydrate Polymers, 94(1), 531–538.
Fatehi, P., Shen, J., Hamdan, F. C., & Ni, Y. (2013). Improving the adsorption of lignocelluloses of prehydrolysis liquor on precipitated calcium carbonate. Carbohydrate Polymers, 92(2), 2103–2110.
Okhrimenko, D. V., Nissenbaum, J., Andersson, M. P., Olsson, M. H. M., & Stipp, S. L. S. (2013). Energies of the adsorption of functional groups to calcium carbonate polymorphs: the importance of -OH and -COOH groups. Langmuir the Acs Journal of Surfaces & Colloids, 29(35), 11062–11073.
Hamdaoui, O., Naffrechoux, E., Suptil, J., & Fachinger, C. (2005). Ultrasonic desorption of p-chlorophenol from granular activated carbon. Chemical Engineering Journal, 106(2), 153–161.
Funding
The research was supported by the National Key R&D Program of China (2017YFD0601001), and the Key Research and Development Program of Jiangsu province in China (BE2015758).
Author information
Authors and Affiliations
Contributions
Jianming Guo and Yong Xu conceived and designed the study. Jianming Guo and Jianglin Zhao performed the experiments and analyzed the data. Jianming Guo prepared the draft manuscript. Yong Xu, Ali Nawaz, Ikram ul Haq, and Wenhuan Chang reviewed and edited the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
The authors declare that they consent to participate.
Consent for Publication
The authors declare that they consent for publication.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Guo, J., Zhao, J., Nawaz, A. et al. In Situ Chemical Locking of Acetates During Xylo-Oligosaccharide Preparation by Lignocellulose Acidolysis. Appl Biochem Biotechnol 193, 2602–2615 (2021). https://doi.org/10.1007/s12010-021-03550-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-021-03550-8