
Abstract
Fermentable sugars are valuable intermediates that can be produced from lignocellulosic biomass, in which hemicellulose and cellulose are favorable candidates for their production. In this chapter, the adaptability of a comprehensive strategy for lignocellulose saccharification is presented. Hemicellulose of reed pole can be hydrolyzed for xylose production by dilute acid first, and then the retained residue from acid hydrolysis, rich in cellulose, can be pyrolyzed for levoglucosan production. Such a process highlighted in this chapter exhibits several attractive features: (1) hemicellulose can be nearly and completely saccharified, giving a xylose yield as high as 92.2 wt%, (2) due to the alteration of chemical composition and physical structure by dilute acid pretreatment, the levoglucosan yield from acid pretreated biomass can be as high as 45.9 wt% as compared with that of un-pretreated reed pole (4.8 wt%), (3) improvement of levoglucosan formation and the inhibition of acetic acid formation from acid pretreated biomass can improve fermentability of its pyrolysate. Furthermore, it is demonstrated that the effect of demineralization, hydrolysis of hemicellulose and improvement of levoglucosan yield is ordered as HCl > H2SO4 > H3PO4.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The dwindling storage of fossil fuels and the increased attention paid to environmental issues have urged numerous researchers to develop the field of bio-refineries. Lignocellulosic biomass, which is the most abundant natural resource, has a positive influence on the renewable energy sector in meeting the growing needs of energy [1]. In general, lignocellulose consists of a massive amount of carbohydrates and predominantly contains various forms of cellulose, hemicellulose, and lignin [2]. Cellulose ((C6H10O5)n) is a class of homopolysaccharide consisting of linear chains of β-D-glucan units connected by β-1,4-glycosidic bonds. Naturally, cellulose can be found in crystalline and amorphous forms. The crystalline cellulose is well organized, tightly bound by inter-chain hydrogen bonds, while amorphous cellulose is less pronounced. Hemicellulose is heteropolysaccharide consisting of hexose (D-mannose, D-galactose, D-glucose) and pentose (D-arabinose, D-xylose). Lignin is a heterogeneous phenylpropanoid polymer, where coumarin alcohol, coniferyl alcohol, and sinapyl alcohol are the primary monomers [3]. Both cellulose and hemicellulose are polysaccharides, which can be converted to ample quantities of fermentable sugars, and they are considered as important intermediates in the conversion of lignocellulose, and can be used as the substrate for microbial fermentation to produce chemicals or biofuels [4]. Saccharification of biomass is one of the pivotal challenges in the development of biofuels [5].
Commonly, enzymatic/acid hydrolysis is the most popular route to depolymerize cellulose into glucose [6]. Enzymatic hydrolysis of cellulose is catalyzed by materials derived from several bacteria and fungi. The simultaneous existences of endo-glucanases, exo-glucanases, and β-glucanases are essential in the enzymatic degradation of cellulose. The endo-glucanases first degrade the cellulose chain to generate free chain-ends. Exo-glucanases subsequently target the sugar chains to isolate cellobiose (dimer of glucose) from the free chain-ends. Eventually, β-glucanases cleave glycosidic bonds in cellobiose to yield glucose. To accomplish the depolymerization of cellulose, each enzyme has a well-defined type of action and all three enzymes show synergistic interactions [7]. There are several advantages for the enzymatic hydrolysis, such as mild reaction conditions, the high selectivity of glucose and no elimination of toxic derivatives in hydrolysate [5]. Overall, enzymatic hydrolysis is a promising approach to selectively converting cellulose into glucose. On the other hand, there are many shortcomings of enzymatic hydrolysis which dramatically restrict its economic benefits and industrial applications, such as long reaction times (ca. days rather than minutes), high costs of enzymes, loss of enzymatic activity due to the inhibition of released glucose, low concentrations of glucose, and the process being vulnerable to contaminants derived from other biomass components [8].
Acid hydrolysis of lignocellulosic biomass, which could date back to the early 19th industrial century and was commercialized in the early twentieth century, has been extensively investigated [9]. Acids can penetrate lignocellulose, decrystallize cellulose and make it more accessible to reactions, leading to the disruption of the network of hydrogen bonds and breakages of glycosidic bonds. Chemicals used for hydrolysis of lignocellulose include mineral acids, such as HF, HCl, H2SO4, H3PO4, as well as organic acids, like fumaric, maleic and oxalic acid. To be effectively performed under atmospheric pressure and at room temperature, a high concentration of acid (e.g. 72 wt% H2SO4) is required. Concentrated acids promote the solubilization of cellulose, and glycosidic bonds are attacked by acid-catalyzed water molecules, followed by hydrolysis of cellulose and hemicellulose to intermediates (e.g. oligosaccharides, monosaccharides). The intermediates are further hydrolyzed to sugars and degraded chemicals (e.g. aldehydes, furfurals, acids). Low energy is consumed in concentrated acid hydrolysis, while the complexities of recycling acid, the risk of disposal of concentrated acid, severe damage of the reactors caused by the strong corrosiveness of acid restrict its large-scale applications [4]. When dilute acids are used instead, H+ leads to the protonation of oxygen atoms of cellulose and activates the glycosidic bonds. Dilute acid is usually performed at higher temperatures (>180 °C) and/or pressures (ca. 1.3 MPa) for minutes to hours. In dilute acids, amorphous cellulose and hemicellulose are prone to hydrolysis, while crystalline cellulose is less prone to be saccharified [10]. Notably, the final concentration of products (e.g. ethanol) is directly proportioned to the initial concentration of the substrate. Thereby, high concentrations of sugars are essential to obtain high ethanol titers to make it easier to distill ethanol from fermentation broth. Nevertheless, the concentration of sugar released from enzymatic/acid hydrolysis is extremely low (approximately 1%) for practical fermentation. It is difficult to overcome the fundamental obstacles and technical limitations of hydrolysis technology [9]. Developing novel efficient and economic strategies for saccharification is warranted.
Despite hydrolysis having attracted much attention, fast pyrolysis is showing a huge potential among researchers aiming towards developing a new road map for lignocellulose saccharification (Fig. 10.1) [1, 11]. Fast pyrolysis, which is a thermo-chemical conversion method, is typically performed in the absence of oxygen at (450–600) °C at very short residence times (ca. seconds), in which lignocellulose thermally decomposes into solid char, non-condensable gas, and pyrolysis oil at the conditions [12]. Pyrolysis oil can contain 75% of the initial energy content of the feed lignocellulose and the bio-oil is typically the desired product [13]. The 1,6-anhydro-β-D-glucopyranose (levoglucosan), derived from cellulose pyrolysis, is the principal component contained in pyrolytic oil [14]. The content of levoglucosan in the pyrolysate is about 80% while the levoglucosan yield obtained from microcrystalline cellulose can reach 70.1 wt% [15]. The feasibility of levoglucosan or pyrolysis oil as a feedstock for eukaryotic and prokaryotic biocatalysts has been demonstrated. The metabolic pathways of levoglucosan have been evaluated for which is understood. Levoglucosan is converted to D-glucose and glucose 6-phosphate in prokaryotic and eukaryotic microorganisms respectively, then metabolized via the glycolytic pathway (Fig. 10.2) [16,17,18]. Levoglucosan can be fermented to citric acid, itaconic acid, malic acid and lipid by Aspergillus terreus, Aspergillus niger CBX-209, Aspergillus oryzae, Rhodotorula glutinis ATCC204091, respectively, giving comparable yields in the glucose fermentation [19,20,21,22]. Furthermore, levoglucosan can be converted to glucose via dilute acid hydrolysis under mild conditions (e.g. 125 °C) [23,24,25]. Many researchers have demonstrated that the process via the hydrolysis of pyrolytic oil of lignocellulose and subsequent fermentation is also feasible [24, 26, 27]. Biochemical studies have confirmed that levoglucosan or its hydrolysate can be metabolized as effectively as glucose. Saccharification of lignocellulose via fast pyrolysis may be an alternative to overcome the barriers identified by hydrolysis conversion. There are several predominant advantages of fast pyrolysis compared with enzymatic or acid hydrolysis of lignocellulose. First, the rate of fast pyrolysis process requires seconds rather than hours or days. Then, fast pyrolysis can be implemented without any catalysts such as expensive enzymes or corrosive acids. Additionally, high concentrations of levoglucosan are easily obtained via fast pyrolysis. Most importantly, economic analyses show that saccharification of lignocellulose by fast pyrolysis has lower operating and capital cost, and improves the economics as compared with enzymatic/acid hydrolysis conversion [28,29,30].

Bio-refining of biomass based on fast pyrolysis (Reprinted with permission from [1], Copyright © 2019 Elsevier)

Nevertheless, pyrolytic depolymerization is hindered by its lack of specificity. Generally, lignocellulosic pyrolysate is a complicated mixture that contains a large number of organic compounds, such as sugars, acids, furans, ketones, aldehydes, and phenols [31]. Levoglucosan yields from cellulose vary widely from 5 C% to 80 C% [32]. However, in the fast pyrolysis of lignocellulose, the yield of levoglucosan (<10 wt% based on cellulose) is much lower as compared with that from cellulose. The supramolecular structure and component of lignocellulose have a profound influence on the formation of levoglucosan [33]. Numerous studies have indicated that the formation of levoglucosan is affected by other components in lignocellulose (e.g. hemicellulose, lignin, ash), especially the alkaline and alkaline earth metals (AAEMs) [34, 35]. Research on fast pyrolysis saccharification still has a lot of unknowns to be explored and requires the investment of considerable effort to achieve high sugar yields. Nevertheless, it is an efficient route to pretreat lignocellulose into favorable raw material to facilitate levoglucosan formation from cellulose fraction [36]. Bio-pretreatment has been employed to alter the structure and chemical composition by microorganisms (e.g. Cunninghamella echinulate, Echinodontium taxodii 2358, Irpex lacteus CD2) [37,38,39,40]. Bio-pretreatment is performed under mild operations at relatively long times (e.g. 25 days) [40]. This pretreatment is environmentally friendly but time-consuming. The improvement in levoglucosan yields is also limited as compared to chemical pretreatments. Hot-water pretreatment, by cooking lignocellulose in water at higher temperatures (180–200 °C) and pressures, is a green hydrothermal reaction. In this process, there exist [H]+ at high concentration and water as an acid-like medium [41]. Due to the removal of part of hemicellulose and lignin, and the removal of ash during the process of hot water washed pretreatment, production of levoglucosan is enhanced, and the quality of bio-oil is improved [42, 43]. Hot-water wash can elevate the yield of levoglucosan to some extent (from 4.9 to 11.7 wt%), but dilute acid pretreatment is more effective (from 4.9 to 23.5 wt%) [43, 44]. Various acid pretreatments (HCl, H2SO4, HNO3 and acetic acid) have been adopted prior to fast pyrolysis, and higher yields of levoglucosan have been obtained [45, 46]. However, acid concentrations higher than a certain threshold (e.g. higher concentration than 3M H2SO4) caused a reduction in levoglucosan yield due to the degradation of cellulose [47]. Among these pretreatments, dilute acid pretreatment is considered as one of the best approaches to improving the yield of levoglucosan. Despite the high expectations for dilute acid pretreatment, pretreatment conditions suitable for levoglucosan formation should be investigated systematically to maximize levoglucosan production from pretreated lignocellulose. Furthermore, the ideal pretreatment process should not only be able to boost levoglucosan production in fast pyrolysis, but also make full utilization of other components and avoid waste disposal. Herein, with the aim of making full utilization of hemicellulose, further maximizing the levoglucosan production, and minimizing the production of inhibitors in fast pyrolysis, various acid pretreatments have been applied before fast pyrolysis of lignocellulose. The alteration of components and structures of lignocellulose before and after pretreatment and the influence of the alteration on the levoglucosan production are investigated.
2 Experimental
2.1 Materials
Reed pole derived from Hunan Province was used as the biomass feedstock. The reed pole was ground from (0.11 to 0.18) mm in diameter, dried to constant weight at 378 K in an oven. Acids (including HCl, H2SO4 and H3PO4) and standard chemicals were purchased from Chuandong Chemical Co., Ltd. (Chongqing) and Sigma (Shanghai).
2.2 Dilute Acid Hydrolysis
Reed pole was hydrolyzed by dilute acid in a high-pressure autoclave. Reed pole (5 g) was pretreated with 50 mL, (0.5 and 5)% HCl, H2SO4 or H3PO4 at 393 K for 1 h. Then, the separation of solid and liquid phases was achieved by filtration. Residual acid in the solid residue was removed with distilled water. Then, the pretreated biomass was dried by lyophilization. The compound contained in the hydrolysate was analyzed by high performance liquid chromatography (HPLC, Waters 2695, Massachusetts, USA) and separated by Bio-Rad Aminex HPX-87H column at 333 K. The content of 5-hydroxymethylfurfural (5-HMF) and furfural was determined using ultraviolet-visible (UV) detector (280 nm), and the content of acetic acid, glucose and xylose was measured using refractive index (RI) detector (323 K). Dilute H2SO4 (5 mmol/L) was applied as the mobile phase at a flow rate of 0.6 mL/min. The chemicals were identified and quantified by the external standard method. The yields of xylose and glucose were defined as below:
where an anhydro correction of 0.88 for xylose and correction of 0.90 for glucose are used to calculate the sugars yields from the corresponding polymers.
2.3 Elemental and Componential Analysis
The content of hydrogen (H), carbon (C) and nitrogen (N) in the reed pole was determined by an elemental analyzer (Vario EL Cube, Hanau, Germany). The content of AAEMs, including potassium (K), sodium (Na), calcium (Ca) and magnesium (Mg) was analyzed by inductively coupled plasma-atomic emission spectrometry (ICP-OES) (Optima 8000, PerkinElmer, Massachusetts, USA). Biomass (0.3 g) was dissolved in 4 mL of concentrated acid mixed by HNO3 and HClO4 (3:1, v/v) before ICP-OES analysis. The composition of carbohydrates was determined by a previous procedure [48]. The un-pretreated and acid pretreated reed pole were hydrolyzed through a two-step H2SO4 hydrolysis, consisting of a 72 wt% H2SO4 hydrolysis at 303 K for 1 h and a 4 wt% H2SO4 hydrolysis at 393 K for 1 h. HPLC was applied to analyze the sugar in the hydrolysate. The content of hemicellulose and cellulose was calculated in light of the content of hexose and pentose.
2.4 Thermogravimetric Analysis
Thermogravimetric analysis was carried out in a TGAQ50 analyzer (TA, New Castle, USA) under a nitrogen atmosphere (20 mL/min) to maintain an inert environment. The heating rate was set at 20 K/min and the samples were initially heated from 323 to 378 K and kept for 10 min at 378 K. Then, the samples were heated to 1023 K. The P (integrated pyrolysis coefficient) was defined as follows:
where DTGmax refers to the maximum rate for decomposition, Ti and Tt refer to the initial and final temperature for decomposition respectively, Tmax refers to the temperature of the maximum rate for decomposition.
The activation energy required for the decomposition of biomass was obtained by numerical calculation with the Distributed Activation Energy Model (DAEM). The typical integral form of DAEM is:
where f(α) is the most probable mechanism function, A is the frequency factor, E is the global activation energy (kJ/mol), T refers to the reaction temperature (K), R is the universal gas constant (8.314 J/(mol·K)).
The fundamental equation of DAEM was defined as:
where Ψ (E, T) is the internal integral:
The probability distribution f(E) was calculated as follows:
2.5 Crystallinity Measurement
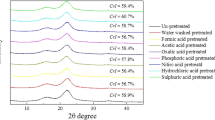
The crystallinity of biomass was scanned by X-ray diffraction (XRD) in X’Pert PROMPD X-ray diffractometer (PANalytical V.B., Almelo, Netherlands). Biomass was scanned by Cu-Kα radiation (λ = 1.54 Å) at a scanning rate of 0.01° per second at a diffraction angle of 5–45°. The crystallinity index (CrI) was calculated by a peak deconvolution method [49].
2.6 Fast Pyrolysis
The process of fast pyrolysis was implemented on a 5200 series CDS pyrolysis probe (CDS analysis, Pennsylvania, USA) connected with Agilent gas chromatograph/mass spectrometer (GC/MS). During each test, biomass (about 200 μg) was weighed with a microbalance (Xp6152, METTLER TOLEDO, Albstadt, Germany) and loaded into the center of a quartz tube with quartz wool plugged on both sides. Biomass was pyrolyzed at 773 K for 20 s at a heating rate of 10 K ms−1. Helium (20 mL/min) was used as the carrier gas continuously passing through the interface to transport the pyrolysis products from the quartz tube to the GC injection port. The split ratio was 50:1. Helium was used as the mobile phase at a flow rate of 1 mL/min in GC. The temperature of the GC oven equipped with an HP-INNO wax capillary column (Agilent 19,091 N-133) was heated from 323 to 503 K at a heating rate of 10 K/min and kept for a residence time of 0.5 h. The mass meter worked in EI mode (70 eV) and the mass scan ranged from m/z of 12–500. The differentiation of compounds was realized by equipping with the NIST mass spectrometry database and quantified by a five-point calibration curve. Each experiment was tested in triplicate. Compound yields were defined as below:
3 Results and Discussion
3.1 Elemental Analysis of Biomass
Elemental analysis of reed pole pretreated by different acids at concentrations of 0.5% and 5% is shown in Table 10.1. No evident change of compositions of C (46.4–49.0 wt%), H (5.8–6.1 wt%) and N (0–0.05 wt%) was observed before and after pretreatment, though there was a slight decrease on N. By comparing the compositions of AAEMs detected from pretreated samples with those from un-pretreated samples, it was found that pretreatment by water or acids could remove AAEMs evidently, especially K+ and Na+, on which both water and acids had a similar effect. For Ca2+ and Mg2+, the contents were decreased after experiencing water-washed, while it was proven that pretreatment by acid had more access to removing Ca2+ and Mg2+. Acid pretreatment could remove 84.1–92.0% of AAEMs. After using a higher concentration of acids, AAEMs could be observed to be further removed. Furthermore, abilities to remove these elements were ordered as HCl > H2SO4 > H3PO4, probably due to the differential concentrations of protons released by acid. Higher concentrations of acid are directly related to the further removal of AAEMs from biomass.
The yield of levoglucosan was sensitive to the content of the initial cellulose, which declined due to a small number of impurities, especially inorganic salts [50]. By interacting with oxygen to change the electronic structure of cellulose and affect the stereochemistry of molecules, AAEMs can favor dehydration, rearrangement reactions, and then fragmentation reaction. Even trace levels of AAEMs naturally found in biomass can shift the pyrolytic pathway, facilitate the formation of lower molecular weight oxygenates while restraining the formation of levoglucosan, by promoting fragmentation rather than depolymerization reaction yielding anhydrosugars [13, 51]. In the research of Patwardhan et al. [51], compared with pure cellulose, when 0.006 mmoles/g NaCl (0.05 wt%) was present in cellulose, the yield of levoglucosan was reduced to 26 wt% from 59 wt%. As the yield of levoglucosan decreased, the yield of low molecular weight compounds increased significantly. As far as the detailed chemical morphology of the pyrolysis products, KCl behaved similarly to NaCl. The participation of 0.004 mmoles KCl/g of cellulose caused a steep decrease in levoglucosan yield (from 59 wt% to 29 wt%). Previous studies had also explored the catalytic role of CaCl2 and MgCl2, which were actually instrumental in the formation of 2-furaldehyde, 5-hydroxymethylfurfural and levoglucosenone, suppressing the formation of levoglucosan [52]. The presence of Ca2+ and Mg2+ resulted in the preliminary formation of char from cellulose, and the conversion of levoglucosan into furans and other lower molecular weight oxygenates [53]. Nevertheless, compared with the former cases of K+ and Na+, the decrease was slightly smaller. Take the yield of levoglucosan into consideration, the order of most mild to strongest influence was Mg2+ < Ca2+ < Na+ < K+ [51, 54]. In this study, cellulose could conveniently be decomposed to levoglucosan without the catalytic effect of AAEMs.
3.2 Componential Analysis of Dilute Acid Hydrolysate and Solid Residual
The amount of hemicellulose and cellulose in reed pole before and after dilute acid hydrolysis, and the main components in dilute acid hydrolysate are analyzed and listed in Tables 10.1 and 10.2, respectively. The un-pretreated sample consisted of 18.9 wt% hemicellulose and 53.1 wt% cellulose. As the water-soluble fraction was released, the content of hemicellulose and cellulose in water-washed biomass increased. Dilute acid hydrolysis was thought to be a favorable approach for the hydrolysis of hemicellulose and amorphous cellulose, as hemicellulose took up 9.5 wt%, 6.3 wt% and 5.2 wt% in biomass pretreated by 0.5% H3PO4, H2SO4 and HCl, respectively. Hemicellulose was proven to disappear absolutely at the residue hydrolyzed by 5% acids regardless of the types of acid. As the principal fraction of hemicellulose, xylan was converted to xylose in acid hydrolysis. The increasing concentration of acid enhanced the formation of xylose. The percent of hemicellulose in the acid hydrolysis residue decreased gradually from 18.9 wt% to 0 wt%. The xylose yield increased from 5.6 to 92.2 wt%. At the acidic concentration of 0.5%, the content of xylose in H3PO4 hydrolysate was 11.8 g/L, less than that 14.7 g/L pretreated by H2SO4 and 15.9 g/L xylose pretreated by HCl. At the acidic concentration of 5.0%, no apparent difference in xylose concentration was observed due to a maximum peak effect achieved at a relatively probable concentration. The content of glucose detected in hydrolysate pretreated differentially was kept in a narrow variation ranging from 2.1 g/L to 4.2 g/L, which was evaluated as a small number compared to xylose, and it could be explained by the harder saccharification of crystalline cellulose than hemicellulose and amorphous cellulose. Acid pretreatment improved the access to removing hemicellulose selectively, with most of the bulk cellulose remaining in the residue. Cellulose took up 75.2 wt%, 78.1 wt% and 78.9 wt% in biomass pretreated by 5% H3PO4, H2SO4 and HCl, respectively. CrI represented the relative ratio of crystalline cellulose contained in the biomass, which was impacted by the presence of amorphous hemicellulose and lignin. Preferential degradation of amorphous hemicellulose and less ordered cellulose by acid hydrolysis, caused the concentration of crystalline cellulose to increase in CrI of pretreated biomass from 56.5% to 62.1% (Fig. 10.3), whereas hydrolysis went towards increasing the formation of byproducts. The most common degradation products were furfural for pentose and 5-HMF for hexose. With increasing acid concentration, particularly, both furfural and 5-HMF, originating from the further decomposition of carbohydrates, were exponentially generated.

XRD patterns of un-pretreated and pretreated biomass
After acid pretreatment, the elimination of hemicellulose and the accumulation of crystalline cellulose could be beneficial to levoglucosan production. There exist some interactions between the components of lignocellulose, dominantly process in pyrolysis. Interactions of cellulose and hemicellulose facilitate the formation of 2,5-diethoxytetrahydrofuran and compounds derived from hemicellulose (e.g. furfural and acetic acid), suppressing the pathway leading to the formation of levoglucosan [55, 56]. Liu et al. [57] also observed that hemicellulose promoted the formation of hydroxyacetaldehyde and suppressed the production of levoglucosan. Previous studies have shown that samples with a higher content of cellulose have the potential to produce more levoglucosan [58]. The crystal allomorph and crystallinity of cellulose could influence the yield of levoglucosan, which was mainly originated from crystalline cellulose [59]. Amorphous samples are easier to be decomposed to a liquid phase than crystalline cellulose to form low molecular weight compounds [60]. Higher yields of levoglucosan can be obtained from cellulosic samples with higher crystallinity [59, 61].
3.3 Thermogravimetric Analysis
The TG and DTG curves of un-pretreated and pretreated biomass are shown in Fig. 10.4. Characteristic parameters of thermal degradation are summarized in Table 10.3. The different relative content of components (hemicellulose, cellulose and lignin) also leads to different pyrolytic profiles of each biomass. Hemicellulose has the strongest reactivity relative to cellulose and lignin, and can be decomposed in a lower temperature range. Cellulose, on the other hand, is the least reactive, requiring higher temperatures to decompose. The thermal decomposition of un-pretreated and water-washed reed pole exhibited two distinct peaks in the DTG curves due to the preferential decomposition of hemicellulose, followed by cellulose. After pretreatment with 5% dilute acid, the first shoulder vanished in the acid pretreated biomass, indicating the removal of hemicellulose. It also confirmed that the profiles of pretreated reed pole had higher Ti, Tmax and Dmax compared with those of un-pretreated sample. The proportion of cellulose in biomass pretreated by H3PO4 was less than that in samples pretreated by H2SO4 or HCl at an acidic concentration of 0.5%. The Tmax matched the differences exactly on their content of cellulose related to the types of acid used in pretreatment, of which the sample pretreated by H3PO4 was 619 K and samples pretreated by H2SO4 or HCl was 626.6 K and 627.7 K, respectively. The severity of pyrolysis was expressed by P. Un-pretreated biomass had the lowest P value (7.8 ×10−6%/K2) compared with the water-washed (10.6×10−6%/K2) and dilute acid pretreated biomass (11.9×10−6 − 13.6×10−6%/K2).

TG (a, c) and DTG curves (b, d) of un-pretreated and pretreated biomass
Table 10.4 shows that the kinetic parameters for the DAEM model, including log10A at 15, 20 and 25. E is the global activation energy, which represents the average activation energy of all reactions. The value of the global activation energy represents the thermal stability of the sample and higher values indicate that the sample is more difficult to decompose. The σ, which is the activation energy deviation, indicates the range of activation energy distribution of a reaction [62]. Smaller activation energy deviations imply more explosive pyrolysis reactions. Each sample could obtain a suitable activation energy density distribution under different pre-exponential factors because of the compensation effect. So, the determination coefficient was above 0.99, which shows that the model agreed well with the experimental curve. Although f(E) depended on the change of the pre-exponential factor, the characters and trends of the parameters reflected were similar. The global activation energy E could be arranged in the following order: un-pretreated < water-washed ≈ dilute acid pretreated sample. The activation energy deviation σ obeyed the following sequence: un-pretreated > water-washed > dilute acid pretreated sample. Removal of hemicellulose and AAEMs, as well as the accumulation of crystalline cellulose, could enhance the thermal stability of biomass during pyrolysis.
It could be seen from the TG curve that the initial volatilizing temperature for lignocellulose containing a great quantity of hemicellulose and AAEMs was lower than the dilute acid pretreated lignocellulose. Similar to the kinetic analysis results, the activation energy required for the decomposition of raw materials with more hemicellulose and AAEMs was the lowest compared with dilute acid pretreated cellulose. Hemicellulose began to decompose at a lower temperature range while cellulose began to decompose at higher temperature intervals. Previous research demonstrated that the presence of AAEMs could lower the initial temperature of lignocellulose degradation. Where K+ is considered as an example. The catalytic influence of K+ has been investigated in previous research [63, 64], where it was shown that K+ could lower the initial and maximum temperature for decomposition, thus reducing the maximum degradation rate and favoring the formation of char. Since hemicellulose and AAEMs dissolved in the acids during the dilute acid pretreatment of lignocellulose, cellulose was the main component decomposed during the subsequent pyrolysis process, resulting in a smaller activation energy deviation and a more explosive reaction. Due to the removal of AAEMs, the formation of biochar in cellulose pyrolysis was also limited. Mr (proportion of pyrolytic residue) analogically explained the weak removal by H3PO4 in hydrolysis, of which the sample pretreated by H3PO4 was 4.7 wt% and samples pretreated by H2SO4 or HCl was 5.9–6.9 wt%. The crystalline structure of biomass is altered during acid pretreatment, which also plays a significant role in the thermal stability of biomass. The crystalline structure affects both the thermal stability and the required activation energy in the thermal decomposition of cellulose [65]. During pyrolysis, amorphous cellulose decomposes more rapidly by heat and degrades at lower temperatures, and substantial heat is absorbed for cleavage of the hydrogen bond network before decomposition [66]. Higher crystallinity of cellulose acts as a barrier for thermal degradation for its packed structure, and intramolecular hydrogen bonds would play important roles in stabilizing the cellulose and suppressing thermal expansion, which improves the thermal stability of cellulose [67]. Thereby, the acid pretreated biomass with higher crystallinity exhibited a higher endothermic activity.
3.4 Fast Pyrolysis
The pyrolysate of lignocellulose is a mixture of various organic compounds, including acids, phenols, furans, ketones, esters, aldehydes and dehydrated sugars [68]. Levoglucosan, which is the target product after pyrolysis, has a sizable proportion in the pyrolysis products. Although a large number of studies have attempted to elaborate on the fast pyrolysis mechanism of the production of levoglucosan from cellulose, there still exists much controversy about the potential generative pathways, mainly including homolytic mechanisms, heterolytic mechanisms and concerted mechanisms [69,70,71]. As shown in Fig. 10.5, the yield of levoglucosan from the un-pretreated sample was very low. The yield of levoglucosan rose from 4.8 wt% without pretreatment to 37.8 wt% after 0.5% dilute acid pretreatment. Abilities to improve the levoglucosan yield were ordered as HCl > H2SO4 > H3PO4. Enhancing the concentration of dilute acid can advance the formation of levoglucosan. The highest levoglucosan yield could be obtained from 5% HCl pretreated biomass (45.9 wt%). The promotion as currently envisioned was mainly because of the demineralization, release of hemicellulose, and the concentration of crystalline cellulose after acid pretreatment. Acetic acid was the major product of the pyrolytic degradation of hemicellulose. Due to the release of hemicellulose by dilute acid hydrolysis, the yield of acetic acid decreased from 5.2 wt% to 0.5 wt%. Levoglucosan has the potential to be used as a favorable feedstock for fermentation to produce biofuels. However, a yield less than its theoretical one from lignocellulose, and the existence of toxic compounds to microorganisms used in fermentation limit application of the pyrolysate [1]. In the fermentation of pyrolysate, carboxylic acids are regarded as some of the most toxic inhibitors for growth and metabolization of biocatalysts [72]. In sum, in the current study, the promotion of levoglucosan production and reduction in the amount of inhibitors formed could improve the fermentability of lignocellulosic pyrolysate. The utilization of dilute acid hydrolysate of biomass for the production of biofuel or biochemistry via microorganism fermentation has been widely demonstrated in previous investigations. Therefore, dilute acid pretreatment could not only saccharify hemicellulose efficiently but also eliminate the negative influence of hemicellulose on the pyrolysis of cellulose.

Yields of levoglucosan and acetic acid from biomass
4 Conclusions and Future Outlook
Herein, the adaptability of an integrated process for lignocellulosic saccharification is provided. The hemicellulose of the reed pole converts to fermentable sugars via dilute acid hydrolysis first. The remaining solid residue from dilute acid hydrolysis, consisting of abundant cellulose, can be utilized to release levoglucosan via fast pyrolysis. Dilute acid hydrolysis as pretreatment could convert most of hemicellulose of biomass and the yield of xylose could reach values as high as 92.2 wt%. The yield of levoglucosan from acid pretreated biomass increased significantly from acid pretreated biomass (45.9 wt%) compared with that of un-pretreated samples (4.8 wt%) due to the demineralization, the release of hemicellulose and the accumulation of crystalline cellulose through dilute acid pretreatment. The abilities to the removal of AAEMs and hemicellulose, and improvement of levoglucosan yield were ordered as HCl > H2SO4 > H3PO4. Further elevating acid concentration from 0.5% to 5% in pretreatment could promote the formation of levoglucosan. The strategy in this research provides a prospective approach for making full use of hemicellulose and cellulose, and obtaining fermentable sugars for a future bio-refinery. Several issues should be considered for further development:
-
1.
Understanding the fundamental mechanisms responsible for levoglucosan formation from cellulose.
-
2.
Illustrating the physical structure features and chemical compositions of lignocellulose in sub-micrometer and nanometer levels and their relationship with levoglucosan formation in fast pyrolysis.
-
3.
Optimizing the process (e.g. suitable feedstock, pretreatment, selective catalysts, pyrolysis temperature, heating rate, residence time and reactor type) to improve levoglucosan yield from biomass and reduce the toxicity of pyrolysate.
-
4.
Developing a multifaceted strategy combining structural, biochemical and metabolic engineering to favor the fermentation of levoglucosan in microorganisms.
References
Jiang LQ, Fang Z, Zhao ZL, Zheng AQ, Wang XB, Li HB. Levoglucosan and its hydrolysates via fast pyrolysis of lignocellulose for microbial biofuels: a state-of-the-art review. Renew Sust Energ Rev. 2019;105:215–9. https://doi.org/10.1016/j.rser.2019.01.055.
Wang SR, Dai GX, Yang HP, Luo ZY. Lignocellulosic biomass pyrolysis mechanism: a state-of-the-art review. Prog Energy Combust Sci. 2017;62:33–86. https://doi.org/10.1016/j.pecs.2017.05.004.
Fang Z. Pretreatment techniques for biofuels and biorefineries. Berlin: Springer; 2012. https://doi.org/10.1007/978-3-642-32735-3.
Guo F, Fang Z, Xu CC, Smith RL Jr. Solid acid mediated hydrolysis of biomass for producing biofuels. Prog Energy Combust Sci. 2012;38(5):672–90. https://doi.org/10.1016/j.pecs.2012.04.001.
Jiang LQ, Zheng AQ, Meng JG, Wang XB, Zhao ZL, Li HB. A comparative investigation of fast pyrolysis with enzymatic hydrolysis for fermentable sugars production from cellulose. Bioresour Technol. 2019;274:281–6. https://doi.org/10.1016/j.biortech.2018.11.098.
Tang S, Dong Q, Fang Z, Miao ZD. Complete recovery of cellulose from rice straw pretreated with ethylene glycol and aluminum chloride from enzymatic hydrolysis. Bioresour Technol. 2019;284:98–104. https://doi.org/10.1016/j.biortech.2019.03.100.
Coseri S. Cellulose: to depolymerize… or not to? Biotechnol Adv. 2017;35:251–66. https://doi.org/10.1016/j.biotechadv.2017.01.002.
Jiang LQ, Zheng AQ, Zhao ZL, He F, Li HB, Wu NN. The comparison of obtaining fermentable sugars from cellulose by enzymatic hydrolysis and fast pyrolysis. Bioresour Technol. 2016;200:8–13. https://doi.org/10.1016/j.biortech.2015.09.096.
Binder JB, Raines RT. Fermentable sugars by chemical hydrolysis of biomass. PNAS. 2010;107:4516–21. https://doi.org/10.1073/pnas.0912073107.
Jiang LQ, Fang Z, Guo F, Yang LB. Production of 2,3-butanediol from acid hydrolysates of Jatropha hulls with Klebsiella Oxytoca. Bioresour Technol. 2012;107:405–10. https://doi.org/10.1016/j.biortech.2011.12.083.
Rover MR, Johnston PA, Jin T, Smith RG, Brown RC, Jarboe L. Production of clean pyrolytic sugars for fermentation. ChemSusChem. 2014;7:1662–8. https://doi.org/10.1002/cssc.201301259.
Bridgwater AV, Meier D, Radlein D. An overview of fast pyrolysis of biomass. Org Geochem. 1999;30(12):1479–93. https://doi.org/10.1016/S0146-6380(99)00120-5.
Carpenter D, Westover TL, Czernik S, Jablonski W. Biomass feedstocks for renewable fuel production: a review of the impacts of feedstock and pretreatment on the yield and product distribution of fast pyrolysis bio-oils and vapors. Green Chem. 2014;16(2):384–406. https://doi.org/10.1039/c3gc41631c.
Lindstrom JK, Proano-Aviles J, Johnston PA, Peterson CA, Stansell JS, Brown RC. Competing reactions limit levoglucosan yield during fast pyrolysis of cellulose. Green Chem. 2019;21:178. https://doi.org/10.1039/c8gc03461c.
Kwon GJ, Kim DY, Kimura S, Kuga S. Rapid-cooling, continuous-feed pyrolyzer for biomass processing preparation of levoglucosan from cellulose and starch. J Anal Appl Pyrolysis. 2007;80(1):1–5. https://doi.org/10.1016/j.jaap.2006.12.012.
Nakahara K, Kitamura Y, Yamagishi Y, Shoun H, Yasui T. Levoglucosan dehydrogenase involved in the assimilation of levoglucosan in Arthrobacter sp. I-552. Biosci Biotechnol Biochem. 1994;58(12):2193–6. https://doi.org/10.1271/bbb.58.2193.
Kitamura Y, Abe Y, Yasui T. Metabolism of levoglucosan (1,6-anhydro-β-D-glucopyranose) in microorganisms. Agric Biol Chem. 1991;55(2):515–21. https://doi.org/10.1080/00021369.1991.10870617.
Yasui T, Kitamura Y, Nakahara K, Abe Y. Metabolism of levoglucosan (1,6-anhydro-β-D-glucopyranose) in bacteria. Agric Biol Chem. 1991;55(7):1927–9. https://doi.org/10.1080/00021369.1991.10870881.
Nakagawa M, Sakai Y, Yasui T. Itaconic acid fermentation of levoglucosan. J Ferment Technol. 1984;62(2):201–3.
Zhuang XL, Zhang HX. Identification, characterization of levoglucosan kinase, and cloning and expression of levoglucosan kinase cDNA from Aspergillus niger CBX-209 in Escherichia coli. Protein Expr Purif. 2002;26(1):71–81. https://doi.org/10.1016/S1046-5928(02)00501-6.
Lian JN, Garcia-Perez M, Chen SL. Fermentation of levoglucosan with oleaginous yeasts for lipid production. Bioresour Technol. 2013;133:183–9. https://doi.org/10.1016/j.biortech.2013.01.031.
Dorsam S, Fesseler J, Gorte O, Hahn T, Zibek S, Syldatk C, Ochsenreither K. Sustainable carbon sources for microbial organic acid production with filamentous fungi. Biotechnol Biofuels. 2017;10(1):242. https://doi.org/10.1186/s13068-017-0930-x.
Prosen EM, Radlein D, Piskorz J, Scott DS, Legge RL. Microbial utilization of levoglucosan in wood pyrolysate as a carbon and energy source. Biotechnol Bioeng. 1993;42(4):538–41. https://doi.org/10.1002/bit.260420419.
Bennett NM, Helle SS, Duff SJB. Extraction and hydrolysis of levoglucosan from pyrolysis oil. Bioresour Technol. 2009;100(23):6059–63. https://doi.org/10.1016/j.biortech.2009.06.067.
Abdilla RM, Rasrendra CB, Heeres HJ. Kinetic studies on the conversion of levoglucosan to glucose in water using Bronsted acids as the catalysts. Ind Eng Chem Res. 2018;57(9):3204–14. https://doi.org/10.1021/acs.iecr.8b00013.
Lian JN, Chen SL, Zhou S, Wang ZH, OʼFallon J, Li CZ, Garcia-Perez M. Separation, hydrolysis and fermentation of pyrolytic sugars to produce ethanol and lipids. Bioresour Technol. 2010;101(24):9688–99. https://doi.org/10.1016/j.biortech.2010.07.071.
Luque L, Oudenhoven S, Westerhof R, van Rossum G, Berruti F, Kersten S, Rehmann L. Comparison of ethanol production from corn cobs and switchgrass following a pyrolysis-based biorefinery approach. Biotechnol Biofuels. 2016;9(1):242. https://doi.org/10.1186/s13068-016-0661-4.
So KS, Brown RC. Economic analysis of selected lignocellulose-to-ethanol conversion technologies. Appl Biochem Biotechnol. 1999;77–79:633–40. https://doi.org/10.1007/978-1-4612-1604-9_57.
Anex RP, Aden A, Kazi KF, Fortman J, Swanson RM, Wright MM, Satrio JA, Brown RC, Daugaard DE, Platon A, Kothandaraman G, Hsu DD, Dutta A. Techno-economic comparison of biomass-to-transportation fuels via pyrolysis, gasification, and biochemical pathways. Fuel. 2010;89:S29–35. https://doi.org/10.1016/j.fuel.2010.07.015.
Zhang YA, Brown TR, Hu GP, Brown RC. Techno-economic analysis of monosaccharide production via fast pyrolysis of lignocellulose. Bioresour Technol. 2013;127(1):358–65. https://doi.org/10.1016/j.biortech.2012.09.070.
Javaid A, Ryan T, Berg G, Pan XM, Vispute T, Bhatia SR, Huber GW, Ford DM. Removal of char particles from fast pyrolysis bio-oil by microfiltration. J Membr Sci. 2010;363(1–2):120–7. https://doi.org/10.1016/j.memsci.2010.07.021.
Maduskar S, Maliekkal V, Neurock M, Dauenhauer PJ. On the yield of levoglucosan from cellulose pyrolysis. ACS Sustain Chem Eng. 2018;6(5):7017–25. https://doi.org/10.1021/acssuschemeng.8b00853.
Dobele G, Meier D, Faix O, Radtke S, Rossinskaja G, Telysheva G. Volatile products of catalytic flash pyrolysis of celluloses. J Anal Appl Pyrolysis. 2001;58:453–63. https://doi.org/10.1016/S0165-2370(00)00128-5.
Zhang J, Choi YS, Yoo CG, Kim TH, Brown RC, Shanks BH. Cellulose-hemicellulose and cellulose-lignin interactions during fast pyrolysis. ACS Sustain Chem Eng. 2015;3:293–301. https://doi.org/10.1021/sc500664.
Nallar M, Wong HW. Hydroxyl group stabilization for increased yields of low-molecular-weight products in the copyrolysis of cellulose and thermoplastics. Ind Eng Chem Res. 2019;58:10776–84. https://doi.org/10.1021/acs.iecr.9b01177.
Wu YX, Jiang LQ, Lin Y, Qian L, Xu FX, Lang XM, Fan SS, Zhao ZL, Li HB. Novel crude glycerol pretreatment for selective saccharification of sugarcane bagasse via fast pyrolysis. Bioresour Technol. 2019;294:122094. https://doi.org/10.1016/j.biortech.2019.122094.
Yang XW, Ma FY, Yu HB, Zhang XY, Chen SL. Effects of biopretreatment of corn stover with white-rot fungus on low-temperature pyrolysis products. Bioresour Technol. 2011;102(3):3498–503. https://doi.org/10.1016/j.biortech.2010.11.021.
Liang JJ, Lin YQ, Wu SB, Liu C, Lei M, Zeng C. Enhancing the quality of bio-oil and selectivity of phenols compounds form pyrolysis of anaerobic digested rice straw. Bioresour Technol. 2015;181:220–3. https://doi.org/10.1016/j.biortech.2015.01.056.
Yu YQ, Zeng YL, Zuo JE, Ma FY, Yang XW, Zhang XY, Wang YJ. Improving the conversion of biomass in catalytic fast pyrolysis via white-rot fungal pretreatment. Bioresour Technol. 2013;134:198–203. https://doi.org/10.1016/j.biortech.2013.01.167.
Wang TP, Ye XN, Yin J, Lu Q, Zheng ZM, Dong CQ. Effects of biopretreatment on pyrolysis behaviors of corn stalk by methanogen. Bioresour Technol. 2014;164:416–9. https://doi.org/10.1016/j.biortech.2014.04.062.
Fang Z, Xu C. Near-critical and supercritical water and their applications for biorefineries. Singapore: Springer; 2014.
Zhurinsh A, Dobele G, Jurkjane V, Meile K, Volperts A, Plavniece A. Impact of hot water pretreatment temperature on the pyrolysis of birch wood. J Anal Appl Pyrolysis. 2017;124:515–22. https://doi.org/10.1016/j.jaap.2017.01.030.
Chandler DS, Resende FLP. Effects of warm water washing on the fast pyrolysis of Arundo Donax. Biomass Bioenergy. 2018;113:65–74. https://doi.org/10.1016/j.biombioe.2018.03.008.
Wang JQ, Wei Q, Zheng JL, Zhu MQ. Effect of pyrolysis conditions on levoglucosan yield from cotton straw and optimization of levoglucosan extraction from bio-oil. J Anal Appl Pyrolysis. 2016;122:294–303. https://doi.org/10.1016/j.jaap.2016.09.013.
Julien S, Chornet E, Overend RP. Influence of acid pretreatment (H2SO4, HCl, HNO3) on reaction selectivity in the vacuum pyrolysis of cellulose. J Anal Appl Pyrolysis. 1993;27(1):25–43. https://doi.org/10.1016/0165-2370(93)80020-Z.
David GF, Justo OR, Perez VH, Garcia-Perez M. Thermochemical conversion of sugarcane bagasse by fast pyrolysis: high yield of levoglucosan production. J Anal Appl Pyrolysis. 2018;133:246–53. https://doi.org/10.1016/j.jaap.2018.03.004.
Kumagai S, Matsuno R, Grause G, Kameda T, Yoshioka T. Enhancement of bio-oil production via pyrolysis of wood biomass by pretreatment with H2SO4. Bioresour Technol. 2015;178:76–82. https://doi.org/10.1016/j.biortech.2014.09.146.
Sluiter A, Hames B, Ruiz R, Scarlata C, Sluiter J, Templeton D, Crocker D. Determination of structural carbohydrates and lignin in biomass. Lab Anal Proce. 2008;1617:1–16.
Park S, Baker JO, Himmel ME, Parilla PA, Johnson DK. Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels. 2010;3:10. https://doi.org/10.1186/1754-6834-3-10.
Kuzhiyil N, Dalluge D, Bai XL, Kim KH, Brown RC. Pyrolytic sugars from cellulosic biomass. ChemSusChem. 2012;5(11):2228–36. https://doi.org/10.1002/cssc.201200341.
Patwardhan PR, Satrio JA, Brown RC, Shanks BH. Influence of inorganic salts on the primary pyrolysis products of cellulose. Bioresour Technol. 2010;101(12):4646–55. https://doi.org/10.1016/j.biortech.2010.01.112.
Liu Q, Wang SR, Luo ZY, Cen KF. Catalysis mechanism study of potassium salts on cellulose pyrolysis by using TGA-FTIR analysis. J Chem Eng Jpn. 2008;41(12):1133–42. https://doi.org/10.1252/jcej.08we056.
Zhu C, Maduskar S, Paulsen AD, Dauenhauer PJ. Alkaline-earth-metal-catalyzed thin-film pyrolysis of cellulose. ChemCatChem. 2016;8(4):818–29. https://doi.org/10.1002/cctc.201501235.
Wang K, Zhang J, Shanks BH, Brown RC. The deleterious effect of inorganic salts on hydrocarbon yields from catalytic pyrolysis of lignocellulosic biomass and its mitigation. Appl Energy. 2015;148:115–20. https://doi.org/10.1016/j.apenergy.2015.03.034.
Wang SR, Guo XJ, Wang KG, Luo ZY. Influence of the interaction of components on the pyrolysis behavior of biomass. J Anal Appl Pyrolysis. 2011;91(1):183–9. https://doi.org/10.1016/j.jaap.2011.02.006.
Wu SL, Sen DK, Hu J, Zhang HY, Xiao R. Cellulose-hemicellulose interactions during fast pyrolysis with different temperatures and mixing methods. Biomass Bioenergy. 2016;95:55–63. https://doi.org/10.1016/j.biombioe.2016.09.015.
Liu QA, Zhao ZP, Wang SR, Luo ZY. Interactions of biomass components during pyrolysis: a TG-FTIR study. J Anal Appl Pyrolysis. 2011;90(2):213–8. https://doi.org/10.1016/j.jaap.2010.12.009.
Miura M, Kaga H, Yoshida T, Ando K. Microwave pyrolysis of cellulosic materials for the production of anhydrosugars. J Wood Sci. 2001;47(6):502–6. https://doi.org/10.1007/BF00767906.
Mukarakate C, Mittal A, Ciesielski PN, Budhi S, Thompson L, Iisa K, Nimlos MR, Donohole BS. Influence of crystal allomorph and crystallinity on the products and behavior of cellulose during fast pyrolysis. ACS Sustain Chem Eng. 2016;4:4662–74. https://doi.org/10.1021/acssuschemeng.6b00812.
Wang ZH, McDonald AG, Westerhof RGM, Kersten SRA, Cuba-Torres CM, Ha S, Pecha B, Garcia-Perez M. Effect of cellulose crystallinity on the formation of liquid intermediate and on product distribution during pyrolysis. J Anal Appl Pyrolysis. 2013;100:56–66. https://doi.org/10.1016/j.jaap.2012.11.017.
Hosoya T, Sakaki S. Levoglucosan formation from crystalline cellulose: importance of a hydrogen bonding network in the reaction. ChemSusChem. 2013;6(12):2356–68. https://doi.org/10.1002/cssc.201300338.
Lin Y, Tian YL, Xia YQ, Fang SW, Liao YF, Yu ZS, Ma XQ. General distributed activation energy model (G-DAEM) on co-pyrolysis kinetics of bagasse and sewage sludge. Bioresour Technol. 2019;273:545–55. https://doi.org/10.1016/j.biortech.2018.11.051.
Le Brech Y, Ghislain T, Leclerc S, Bouroukba M, Delmotte L, Brosse N, Snape C, Chaimbault P, Dufour A. Effect of potassium on the mechanisms of biomass pyrolysis studied using complementary analytical techniques. ChemSusChem. 2016;9(8):863–72. https://doi.org/10.1002/cssc.201501560.
Zhang LQ, Li SS, Ding HZ, Zhu XF. Two-step pyrolysis of corncob for value-added chemicals and high-quality bio-oil: effects of alkali and alkaline earth metals. Waste Manag. 2019;87:709–18. https://doi.org/10.1016/j.wasman.2019.03.002.
Poletto M, Pistor V, Zeni M, Zattera AJ. Crystalline properties and decomposition kinetics of cellulose fibers in wood pulp obtained by two pulping processes. Polym Degrad Stab. 2011;96(4):679–85. https://doi.org/10.1016/j.polymdegradstab.2010.12.007.
Patai S, Halper Y. Pyrolytic reaction of carbohydrates.9. Effect of additive of thermal behavior of cellulose samples of different crystallinity. Isr J Chem. 1970;8:655–62. https://doi.org/10.1002/ijch.197000079.
Poletto M, Ornaghi HL, Zattera AJ. Native cellulose: structure, characterization and thermal properties. Materials. 2014;7(9):6105–19. https://doi.org/10.3390/ma7096105.
Lu Q, Hu B, Zhang ZX, Wu YT, Cui MS, Liu DJ, Dong CQ, Yang YP. Mechanism of cellulose fast pyrolysis: the role of characteristic chain ends and dehydrated units. Combust Flame. 2018;198:267–77. https://doi.org/10.1016/j.combustflame.2018.09.025.
Shen D, Gu S. The mechanism for thermal decomposition of cellulose and its main products. Bioresour Technol. 2009;100(24):6496–504. https://doi.org/10.1016/j.biortech.2009.06.095.
Ponder GR, Richards GN, Stevenson TT. Influence of linkage position and orientation in pyrolysis of polysaccharides: a study of several glucans. J Anal Appl Pyrolysis. 1992;22(3):217–29. https://doi.org/10.1016/0165-2370(92)85015-D.
Mayes HB, Broadbelt LJ. Unraveling the reactions that unravel cellulose. J Phys Chem A. 2012;116(26):7098–106. https://doi.org/10.1021/jp300405x.
Lian JN, McKenna R, Rover MR, Nielsen DR, Wen ZY, Jarboe LR. Production of biorenewable styrene: utilization of biomass-derived sugars and insights into toxicity. J Ind Microbiol Biotechnol. 2016;43(5):595–604. https://doi.org/10.1007/s10295-016-1734-x.
Acknowledgements
Financial support provided by the National Natural Science Foundation of China (No. 51606204, 51876208), the Project Foundation of Guangdong province and Guangzhou city (No. 2017A020216007 and No. 201707010236), the Open Project of State Key Laboratory of Microbial Technology (No. M2019-10) are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Jiang, Lq., Wang, Xb., Zhao, Zl., Li, Hb. (2020). Levoglucosan Production by Fast Pyrolysis of Biomass After Dilute Acid Pretreatment. In: Fang, Z., Smith Jr, R.L., Xu, L. (eds) Production of Biofuels and Chemicals with Pyrolysis. Biofuels and Biorefineries, vol 10. Springer, Singapore. https://doi.org/10.1007/978-981-15-2732-6_10
Download citation
DOI: https://doi.org/10.1007/978-981-15-2732-6_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-2731-9
Online ISBN: 978-981-15-2732-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)