
Abstract
Withania somnifera, popularly known as Indian ginseng, is one of the most important medicinal plants. The plant is well studied in terms of its pharmaceutical activities and genes involved in biosynthetic pathways. However, not much is known about the regulatory mechanism of genes responsible for the production of secondary metabolites. The idea was to identify miRNA transcriptome responsible for the regulation of withanolide biosynthesis, specifically of root and leaf tissues individually. The transcriptome data of in vitro culture of root and leaf tissues of the plant was considered for miRNA identification. A total of 24 and 39 miRNA families were identified in root and leaf tissues, respectively. Out of these, 15 and 27 miRNA families have shown their involvement in different biological functions in root and leaf tissues, respectively. We report here, specific miRNAs and their corresponding target genes for corresponding root and leaf tissues. The target genes have also been analyzed for their role in withanolide metabolism. Endogenous root-miR5140, root-miR159, leaf-miR477, and leaf-miR530 were reported for regulation of withanolide biosynthesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Withania somnifera is one of the most reputed medicinal plants of solanaceae family due to its diverse medicinal properties [1]. Till date, a lot of work has been carried out in terms of the medicinal uses of withanolides for their anti-inflammatory, anti-stress, anti-tumor, antioxidant, anti-aging, immunomodulatory, immunoprophylactic, rejuvenating, and aphrodisiac activities [2, 3]. The pharmacological activities of different withanolides have also been reported for various cognitive and neurological disorders such as Parkinson’s and Alzheimer’s disease, intestinal disorders, and hepatoprotection [4]. Withanolides are comprised of withaferin A, withanolide A, and withanone. These withanolides are produced by different parts of the plant (i.e., root, leaf, stem and bark) independently rather than being imported from leaves [5]. The content of withaferin A is reported to be highest in leaf followed by bark, stem, and root whereas withanolide A is biosynthesized majorly in roots [3, 6]. Therefore, both leaf as well as root are important plant parts for the production of withanolides. The biosynthesis of these metabolites is dependent on the genes associated with corresponding metabolic pathways. The changes in the expression pattern of these genes and enzymes are reported to be regulated by different biomolecules such as transcription factor proteins and non-coding RNAs. The transcription factors play an important role in the regulation of responsible genes through binding at their regulatory regions. The mechanism of these transcription factors is further regulated by specific type of non-coding RNAs. These non-coding RNAs are not translated into protein but have their significance as regulatory elements. Non coding RNAs consist of different members of varied sizes and mode of action. One of these non-coding RNAs are microRNAs. miRNAs are small (20–22 bp), endogenous and show negative modulation of the expression of genes. The miRNAs are reported to regulate the expression of genes through inhibition of translation and promoting the degradation or cleavage of the target genes. It is reported that mature miRNAs are highly conserved throughout the plant kingdom [7]. Usually animal and plant miRNA biogenesis is almost similar but at some places they show major differences, which distinguishes between them: (1) Plant precursor miRNAs have larger and more variable stem-loop structurtes as compared to animal, (2) plant mature miRNAs pair their target sites with near-perfect complementarity, (3) in animals, miRNAs mainly recognize several target sites in 3’ UTR of mRNAs and inhibit translation, while plant miRNAs often recognize a single target site and cleave the mRNA [8]. Different approaches have been used for miRNA identification such as comparative genomics approach and NGS technology used for novel miRNA and homology-based conserved miRNA identification [9]. Plant miRNAs are involved in different biological processes [10] such as, auxin responses [11], stress responses [12], floral identity [13], and leaf polarity [14]. One of the biological functions of miRNA is to regulate secondary metabolite synthesizes in plants [15]. In the present study, we have carried out a comprehensive in silico analysis to identify miRNA transcriptome in vitro cultured W. somnifera leaf and root tissues. Here, we have used homology approach for the identification of miRNAs in W. somnifera transcriptome of leaf and root tissues. Different bioinformatics tools were used for the identification of miRNAs and their corresponding targets. Functional annotation of putative targets, biological network analysis, and pathway analysis were also performed. Through RT-PCR, differential gene expression analysis was also performed for the validation of identified miRNAs and their targets involved in withanolide biosynthesis pathway. To best of our knowledge, it is the first study on identification of miRNAs in W. somnifera.
Materials and Methods
Withania somnifera Root and Leaf Transcriptome Dataset
The transcriptome sequences used in this study for identifying conserved miRNAs and their targets have recently been sequenced, assembled, annotated and deposited in Short Read Archive (SRA) of NCBI with accession number SRP040231 [16]. In brief, the in vitro cultures of adventitious root and leaf tissues of W. somnifera were subjected to paired-end (PE) sequencing with Illumina platform. Through sequencing, 135,186,223 and 113,849,837 reads were identified from root and leaf tissues, respectively. The low-quality reads and primer adapters have already been removed using NGS QC Toolkit [17]. The sequence assembly was done for both the tissue specific data through SeqManNGen module of DNAstar (http://www.dnastar.com/t-nextgen-seqman-ngen.aspx).
Prediction of Root and Leaf Pre-miRNAs
For the prediction of pre-miRNAs, the reference dataset on experimentally identified miRNA sequences were obtained from the plant miRNA database, miRBase (http://www.mirbase.org/) [18]. This database contains 28,645 published hairpin precursor miRNA sequences and 35,828 mature miRNAs with associated annotations from approximately 223 plant species.The similarity search was performed between the assembled sequences and known miRNA sequences using BLASTN program of C-mii tool [19]. The putative miRNA candidates were scanned against the reported miRNAs in miRBase using BLASTN (e-value cut-off: 10), BLASTX (evalue b = 1e−5) was performed against protein-databases. UniprotKB/Swissprot (release 2010_12) and UniProtKB/TrEMBL (release 2011_01) were used to remove the protein-coding sequences. RNA database Rfam 10 was used to predict structures of primary and precursor miRNAs. UNAFold was used with the parameters of maximum base pair distance = 3000, maximum bulge/interior loop size 30, and single tread run of 37 °C temperature [9].
Prediction of Mature miRNAs
Homology search-based miRNA identification was performed using following criteria: (1) The length of the predicted mature miRNAs should be 19–25 nucleotides; (2) maximum four mismatches were allowed for the predicted mature miRNAs against reference miRNA; (3) localization of the mature miRNA within stem–loop structure should be one arm; (4) not more than 5 mismatches were allowed between miRNA sequences and the guide miRNA sequence; (5) A + U content should be high; and (6) minimal folding free energy (MFE) and MFE index (MFEI) value of the secondary structure should be highly negative.
Prediction of miRNA Targets
The miRNA targets were identified to understand the biological functions of the corresponding miRNAs. The miRNA target sequences were predicted on the basis of their perfect or nearly perfect complementarity with mature miRNA sequences. The identified miRNA candidates were used for the target identification against the tissue specific transcript. Following criteria were set for the prediction of miRNA target genes: (1) not more than four mismatches were allowed between predicted miRNAs and target gene; (2) no mismatches were allowed for 10th and 11th positions of complementary site as it is considered as a cleavage site; and (3) maximum 4 GU pair was allowed in the complimentary alignment [9].
Filtering of Tissue-Specific miRNA and Their Targets
After prediction of miRNA and their putative targets, we have filtered root and leaf-specific miRNAs and their targets and also filtered common miRNAs and targets identified in both tissues using MATLAB software (https://www.mathworks.com/products/matlab.html).
Functional Annotation, Network and Pathway Analysis of Putative miRNA Targets Root and Leaf Tissues
To annotate all putative targets, BLASTX-based approach was used to compare the sequences to nr database downloaded from NCBI (http://www.ncbi.nlm.nih.gov/). The GI identifiers of the best BLASTX, for all hits having an evalue e−9 and a degree of similarity 70%, were mapped in the UniprotKB protein database (http://www.uniprot.org/). Finally, the UniprotKB accessions were used to extract gene ontology terms for further functional annotations. Blast2GO software v1.3.3 (http://www.blast2go.org) [20] was used to perform basic statistics on GO annotations as reported earlier [21, 22]. To understand the function of W. somnifera miRNAs and their regulatory targets, the biological network and pathway analysis was performed through Cytoscape [23] and KAAS server, respectively [24].
Expression Analysis of Identified miRNAs and Their Targets Involved in Withanolide Biosynthesis of Root and Leaf Tissues
Total RNA was isolated from leaf and root tissues of three independent replicates of control and drought-treated plants of W. somnifera [25]. The RT-PCR was done in three replicates with 1 μL cDNA using the SYBR Green Master Mix (Applied Biosystems) with an ABI 7900HT RT-PCR detection system as prescribed in the manufacturer’s protocol (Applied Biosystems). Each reaction mixture consisted of 2 μl cDNA, 15 μl SYBR green mix (2X) (TaKaRa), 2 μl (5 pmol/μl) of both forward and reverse primers, and 11 μl PCR-grade water equating to a final volume of 30 μl. This reaction mix was dispensed in a 384-well PCR plate in triplicates. The thermal profile of the reaction was an initial denaturation at 95 °C for 2 min, followed by 40 cycles at 95 °C for 10 s and 60 °C for 10 s. Quantification of transcript (mRNA expression) levels was carried out by using the CT quantitative methods. Normalization was carried out by subtracting CT value of ubiquitin from CT value of target gene. Following normalization, the relative abundance of transcripts was calculated as RQ = 2−ΔΔCT [25]. The gene-specific and ubiquitin (internal control) primers were designed by PRIMER3 (http://frodo.wi.mit.edu/primer3/input.htm). The primer details are listed in Supplementary Table 1.
Results
Identification of Conserved miRNA Families in W. somnifera Transcriptome
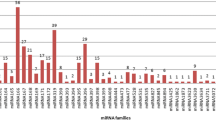
Through assembly of reads from root as well as leaf tissue, 14,405 and 13,289 contigs were identified, respectively. Out of these contigs, 10,637 and 9498 were considered further for miRNA analysis through C-mii tool. Various experimentally identified miRNAs available in miRBase database were considered for miRNA identification in W. somnifera. The homology and structure similarity based searches were considered for the identification of mature miRNAs. In root tissues, we have identified 24 miRNA families from 10,637 contigs. We observed pre- miRNA MFEI range from − 0.60 to − 1.21 kcal/mol with an average − 0.79 kcal/mol and MFE of pre- miRNA varied from − 9.9 to − 281.9 kcal/mol with an average − 45.56 kcal/mol. Mature miRNA length varied from 18 to 24 nt with an average 20 nt. AU and GC content of predicted mature miRNAs families was in the range of 42.11 to 71.43 and 33.33 to 57.89 with an average of 55.33 and 44.66, respectively (Fig. 1a). In leaf tissue, 39 miRNA families were identified from 9498 contigs. Pre- miRNA MFEI ranged from − 0.60 to − 1.42 kcal/mol with an average − 0.71 kcal/mol and MFE of pre- miRNA from − 9.2 to − 228 kcal/mol with an average − 39.69 kcal/mol was obsereved. Mature miRNA length varied from 18 to 22 nt with an average 20 nt. AU and GC content of predicted mature miRNAs families was in the range of 23.81 to 80.75 and 19.05 to 76.19 with an average of 58.38 and 41.61, respectively (Supplementary Table 2, Fig. 1b).

Predicted mature miRNA families a root tissue and b leaf tissue
Co-expressed miRNAs in Root and Leaf Tissues of Withania somnifera
Sixteen miRNA families (miR5565, miR1132, miR156, miR5140, miR473, miR5381, miR1109, miR5389, miR5139, miR1096, miR1134, miR1097, miR5137, miR319, miR1088 and miR948) were expressed only in root tissue. In leaf tissue, 31 miRNA families (miR5652, miR396, miR5631, miR5246, miR1078, miR4993, miR5262, miR3979, miR394, miR1534, miR825, miR444, miR2088, miR947, miR395, miR5658, miR477, miR1852, miR4996, miR2105, miR1521, miR902, miR866, miR1426, miR1847, miR2654, miR1075, miR5632, miR530, miR5079 and miR1031) were expressed. Coexpression analysis of predicted miRNA families revealed that eight miRNA families (miR164, miR169, miR159, miR398, miR476, miR5085, miR172 and miR5303) were expressed in both root and leaf tissues (Fig. 2).

Distribution of miRNA families in root and leaf tissues
Analysis of Endogenous Putative miRNA Targets and Their Biological Networks in Root and Leaf Tissues
The identification of targets for corresponding miRNAs was done using the tissue specific data. Out of 24 predicted miRNA families in root tissue, 15 were identified to be involved in the regulation of 128 putatve targets in root (Fig. 3a). Whereas, out of 39 miRNA families in leaf tissue, 27 were involved in the regulation of 126 putative targets in leaf (Fig. 3b). The miRNA families coexpressed in both root and leaf tissues, 6 and 5 miRNA families were involved in the regulation of 58 and 37 putative targets, respectively (Fig. 3c). Network analysis of predicted miRNA families of root and leaf tissue revealed that most of the single miRNA families may regulate multiple putative targets and single putative tagets are regulated by multiple miRNA families (Supplementary Fig. 1a and 1b, Supplementary Fig. 2a and 2b).

Predicted miRNA families putative targets a only root, b only leaf, and c root and leaf
Role of Predicted miRNA Families in Different Pathways of Root and Leaf Tissues
Pathway analysis of predicted miRNA families in root and leaf tissue revealed that five miRNA families were involved in the regulation of 18 biosynthetic pathways in root tissues (Supplementary Fig. 3a). In leaf, 16 miRNA families were involved in the regulation of 75 different pathways (Supplementary Fig. 4a). Pathway analysis was also done for coexpressed miRNA families. The analysis revealed that out of 8 coexpressed miRNA families, 6 from root tissue were involved in the regulation of 18 different biosynthetic pathways (Supplementary Fig. 3b) whereas in leaf tissues, 5 miRNA families were involved in regulation of 30 different pathways (Supplementary Fig. 4b).
Regulation of Secondary Metabolite Biosynthesis by Predicted miRNA Families of Root and Leaf Tissue
Our results showed that miR5140 identified only in root tissues was involved in the regulation of steriod biosynthesis-related gene (Supplementary Table 3, Supplementary Fig. 3a). miR159, miR172 and miR5303 which were identified both in root and leaf tissues were involved in the regulation of steroid, stilbenoid, diarylheptanoid and gingerol biosynthesis, phenylpropanoid biosynthesis-related genes (Supplementary Table 3, Supplementary Fig. 3b and 4b). In leaf tissue, we found that out of predicted miRNA families, miR5658, miR477, miR1426, miR530 and miR5079 were involved in the regulation of isoquinoline biosynthesis, glycosyltransferases, ubiquinone and other terpnoid-quinone biosynthesis, phenylpropanoid biosynthesis, phenylpropanoid biosynthesis, carotenoid biosynthesis, diterpenoid biosynthesis-related genes, respectively (Supplementary Table 3, Supplementary Fig. 4a).
Analysis of miRNA Families Involved in Withanolide Biosynthesis
Analysis revealed that some miRNA families identified from root and leaf tissues were involved in withanoides biosynthesis. Two miRNA families in root (miR5140 and miR159) were involved in the regulation of cycloartenol synthase (CAS1) and sterol delta-7 reductase 1 (DWF1) genes, respectively whereas three miRNA families in leaf (miR5079, miR530 and miR477) were involved in the regulation of CYP82G, secoisolariciresinol dehydrogenase (ABA2) and zeatin o-glycosyl transferase (UGTs) genes, respectively (Supplementary Table 3, Fig. 4).

Schematic diagram of withanolide biosynthesis with involved predicted miRNA families of root (orange circle) and leaf tissue (green circle)
Expression Analysis of Putative Root and Leaf Tissues miRNAs and Their Targets Involved in Withanolide Biosynthesis
In this study, RT-PCR was used to investigate the expression pattern of withanolide biosynthesis responsive miRNAs and their putative targets in W. somnifera root and leaf tissues. Root-miR5140 and root-miR159 targets CAS1 and DWF1 genes, respectively. Here, we have analyzed the expression of root-miR5140 with target gene CAS1 and root-miR159 with target gene DWF1 under control vs. severe drought stress condition. Results showed that differential expression of both root tissue miRNAs with their targets were down regulated in severe stress condtion (Fig. 5a). DWF1 gene showed comparatively higher expression than root-miRNA159. All the miRNAs identified from leaf specific dataset were upregulated under drought stress. Their expression were increased along with their target genes under increasing drought stress (Fig. 5b–d). All the studied three miRNA showed hightest upregulation at the 12th day of drought after that it was decreased. Leaf-miR477 and leaf-miR530 targets UGT and ABA2 genes, respectively. Expression of leaf-miR477 and leaf-miR530 was increased by 480 and 4.3 times in comparison to watered plants at the 12th day of drought. However, their corresponding target genes were upregulated by 1.6 and 2 fold at 12th day of drought (Fig. 5b, c). The expression of leaf-miR159 was increased by 336.82 fold along with 22 times upregulation of its target DWF1gene (Fig. 5c).

Differential gene expression of miRNAs in root (a) and leaf tissue (b, c, d) under different drought conditions
Discussion
Withania somnifera (ashwagandha), a multipurpose medicinal plant is a rich reservoir of pharmaceutically active compounds. This plant has been included in the list of top 32 prime concerned medicinal plants by the National Medicinal Plant Board of India (http://www.nmpb.nic.in) owing to its huge demand in both domestic and international markets [26]. All parts of ashwagandha like leaf, stem, flower, root, seed and bark are used as pharmaceutical constituents [27]. Various chemical constituents like withanolides, alkaloids, sitoindosides, flavanoids, saponins, glycosides and triterpenoids present in W. somnifera are important due to their applications in many industrial products [6, 28]. The numerous therapeutic applications of W. somnifera are related to anti-inflammatory activities, action on the immune system [29, 30], circulatory system [31], diabetes [32], central nervous system [33, 34] and anticancer activity [35]. Presently, thousands of miRNA families have been identified in diverse species of different plant families. But still needs to be explored a large number of plant miRNAs and their regulation in different biological function [36]. To gain the new insights into the regulatory role of miRNAs in withanolide metabolism, we identified the miRNAs from the transcriptome data of root and leaf tissues. Many reports are available for the identification and characterization of miRNAs in the transcriptome data such as Typha angustifolia in response to cadmium stress [37], steviol glycosides biosynthetic pathway of Stevia rebaudiana [38], alkaloid biosynthesis of opimum poppy [15], carotenoid biosynthesis of tomato [39], regulation of phenylpropanoid pathway in Arabidopsis [40]. Despite the accumulated knowledge of the microRNA-mediated regulation of several processes, the involvement of miRNA families in regulating secondary metabolies biosynthesis of pharmaceutically and commercially important plants is still poorly understood [41]. In present study, 24 and 39 miRNA families were identified from root and leaf tissue, respectively (Supplementary Table 2, Fig. 1a and b). The average mature miRNAs length, AU and GC content, pre-miRNAs MFEI and MFE in root tissue were 20 nt, 55.33, 44.66, − 0.79 kcal/mol and − 45.56 kcal/mol, respectively. On the other hand in leaf tissue average mature miRNAs length, AU and GC content, pre-miRNAs MFEI and MFE were 20 nt, 19.05, 76.19, − 0.71 kcal/mol and − 3969 kcal/mol, respectively. The above reported characteristics of miRNA genes were in agreement with many previous studies ([9, 42,43,44,45,46], B. H. [47]). Coexpression analysis of miRNA revealed that eight miRNA families (miR164,miR169, miR159, miR398, miR476, miR5085, miR172 and miR5303) were expressed in both root and leaf tissues. Target prediction revealed miRNAs that are present only in root and leaf and coexpressed miRNAs regulation in both tissues were observed in different biological processes (Supplementary Tables 4 and 5). In this study, our main focus was to identify miRNAs involved in the regulation of genes related to secondary metabolic pathways, particularly withanolide biosynthesis. Pathway analysis revealed that four miRNA families (miR5140, miR159, miR172 and miR5303) identified in root tissues were involved in the regulation of secondary metabolite biosynthesis (Supplementary Table 3). Out of these four miRNA families, three (miR159, miR172 and miR5303) were expressed in both root and leaf tissues. But these three miRNAs (miR159, miR172 and miR5303) were found to target the genes involved in regulation of secondary metabolites only in root tissue. Earlier reports are available on these four miRNAs from the roots of W. somnifera viz., miR5140, involved in magnesium transporter CorA-like family protein [48], miR159 involved in regulation of GAMYB like genes [49], NACs [50]. miR172 targets the genes which lead to the biosynthesis and accumulation of lycopene [39] and main candidates acting as mobile signals for tuberization [51]. miR5303 targeting protein for Xklp2 (TPX2)/Mpp10 domain containing protein [52] and also responsible for amino peptidase activity [53]. In case of leaf tissue, four miRNAs (miR477, miR1426, miR530 and miR5079) which express only in leaf, target the secondary metabolite biosynthesis-related genes (Supplementary Table 3). Many previous reports are available on these four miRNAs found in leaf of W. somniferavz., viz., mitogen-activated protein kinase, heat shock cognate protein, cytosolic ascorbate peroxidase myo-inositol-1-phosphate synthase, dual specificity protein phosphatase, glutamate synthase-related genes regulate by miR477[54]. miR1426 showed preferential expression at different development stages of leaves and flowers as reported earlier [55]. Regulation of anther development and male sterility in 7B-1 male-sterile tomato mutant [56] and targeting the argonaute 1 gene indicated a second autoregulatory mechanism for miRNA regulation by miR530 [57]. miR5079 was reported to express preferentially in the tolerant rice genotype N22 at high temperature [58]. Analysis of miRNAs families which were involved in regulation of withanolides biosynthesis-related genes revealed that miR5140 and miR159 of root tissue target CAS1 and DWF1 genes respectively which play an important role in wihanolides biosynthesis. CAS1 has been reported as central intermediate in the metabolic route toward withanloide biosynthesis. Differential expression of root-miR5140 and root-miR159 and their corresponding target genes were downregulated. Hence, these miRNAs were positively regulating their targtes (Fig. 5a). The upregulation of leaf-miR477, leaf-miR530 and leaf-miR159 showed the negative regulation of their corresponding targets at 12th day drought, specifically (Fig. 5b, c and d). DWF1 was also found as multifunctional enzyme which may carry out isomerization and reduction of post 24-methylene cholestrol intermediates in withanolide biosynthesis [27]. Study of leaf tissue revealed the involvement of miR477 and miR530 in withanoide biosynthesis by targeting UGTs and ABA2, respectively. In earlier study [59], it was reported that plant family 1 UGTs catalyze the glycosylation of surplus bioactive natural compounds. This is frequently the concluding step for biosynthesis of various natural products [60], which lead to enhancement of withanolides [27]. ABA2 is required for the plastidal MEP pathway synthesis, IPP (isopentenylnpyrophosphate) and DMAPP (dimethyl allyl pyrophosphate) [27]. Dual autonomous pathways for the isoprenoid precursor biosynthesis co-exist in plant cell including the classical cytosolic mevalonic acid (MVA) pathway and the alternative route, plastidial methylerythritol phosphate (MEP) pathway [61]. Triterpenoid backbone, like other terpenoid compounds is biosynthesized by metabolic pathway requiring isoprene units (IPP and DMAPP) as precursors. Therefore, isoprenogenesis could be one of the key upstream metabolic processes governing flux of isoprene units for synthesis of metabolic intermediate(s) of triterpenoid pathway committed to withanolide biosynthesis [27].
Conclusion
Transcriptome analysis of W. somnifera for prediction of miRNA identification and characterization showed that miR5140, miR159, miR172 and miR5303 of root tissue and miR477, miR1426, miR530 and miR5079 of leaf tissue were involved in regulation of secondary metabolites. Endogenous root-miR5140, root-miR159, leaf-miR477 and leaf-miR530 may be helpful for, increasing the withanoides yield. In addition, root-miR159, root-miR172 and leaf-miR530 were endogenously involved in regulation of secondary metabolite-related mRNAs. Several studies have reported the involvement of plant miR172, miR530 and miR159 in the regulation of crosskingdom (exogenous) mRNAs [62,63,64]. These predicted miRNA families of W. somnifera may also be further explored for regulation of human disease-related mRNA. This study will help to further understanding W. somnifera miRNA-based genetic regulation of different biosynthetic pathways.
References
Kaileh, M., VandenBerghe, W., Boone, E., Essawi, T., & Haegeman, G. (2007). Screening of indigenous Palestinian medicinal plants for potential anti-inflammatory and cytotoxic activity. Journal of Ethnopharmacology, 113, 510–516.
Mirjalili, M. H., Moyano, E., Bonfill, M., Cusido, R. M., & Palazon, J. (2009). Steroidal lactones from Withania somnifera, an ancient plant for novel medicine. Molecules, 14(7), 2373–2393. https://doi.org/10.3390/molecules14072373
Siriwardane, A. S., Dharmadasa, R. M., & Samarasinghe, K. (2013). Distribution of withaferin A, an anticancer potential agent, in different parts of two varieties of Withania somnifera (L.) Dunal. grown in Sri Lanka. Pak J BiolSci, 16, 141–144.
Verma, S. K., & Kumar, A. (2011). Therapeutic uses of Withania somnifera (ashwagandha) with a note on withanolides and its pharmacological actions. Asian Journal of Pharmaceutical and Clinical Research, 4, 1–4.
Sangwan, R. S., Chaurasiya, N. D., Sangwan, P. L., Misra, L. N., Tuli, R., & Sangwan, N. S. (2008). Withanolide A is inherently de novo biosynthesized in roots of the medicinal plant Ashwagandha (Withania somnifera). Plant Physiology, 133(2), 278–287. https://doi.org/10.1111/j.1399-3054.2008.01076.x
Pal, S., Singh, S., Ashutosh, K. S., Madan, M. G., Suman, P. S. K., & Ajit, K. S. (2011). Comparative withanolide profiles, gene isolation, and differential gene expression in the leaves and roots of Withania somnifera. The Journal of Horticultural Science and Biotechnology, 86(4), 391–397. https://doi.org/10.1080/14620316.2011.11512779
Da Silva, A. C., Grativol, C., Thiebaut, F., Hemerly, A. S., & Ferreira, P. C. G. (2016). Computational identification and comparative analysis of miRNA precursors in three palm species. Planta, 243(5), 1265–1277. https://doi.org/10.1007/s00425-016-2486-6
Yang, T., Xue, L., & An, L. (2007). Functional diversity of miRNA in plants. Plant Science, 172(3), 423–432. https://doi.org/10.1016/j.plantsci.2006.10.009
Srivastava, S., Singh, N., Srivastava, G., & Sharma, A. (2017). miRNA mediated gene regulatory network analysis of Cichoriumintybus (chicory). Agri Gene, 3, 37–45. https://doi.org/10.1016/j.aggene.2016.11.003
Mallory, A. C., & Vaucheret, H. (2006). Functions of microRNAs and related small RNAs in plants. Nature Genetics, 38(Suppl), S31–S36. https://doi.org/10.1038/ng1791
Wang, J.-W., Wang, L.-J., Mao, Y.-B., Cai, W.-J., Xue, H.-W., & Chen, X.-Y. (2005). Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis. Plant Cell, 17(8), 2204–2216. https://doi.org/10.1105/tpc.105.033076
Jones-Rhoades, M. W., & Bartel, D. P. (2004). Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Molecular Cell, 14(6), 787–799. https://doi.org/10.1016/j.molcel.2004.05.027
Chen, X. (2004). A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science, 303(5666), 2022–2025. https://doi.org/10.1126/science.1088060
Palatnik, J. F., Allen, E., Wu, X., Schommer, C., Schwab, R., Carrington, J. C., & Weigel, D. (2003). Control of leaf morphogenesis by microRNAs. Nature, 425(6955), 257–263. https://doi.org/10.1038/nature01958
Boke, H., Ozhuner, E., Turktas, M., Parmaksiz, I., Ozcan, S., & Unver, T. (2015). Regulation of the alkaloid biosynthesis by miRNA in opium poppy. Plant Biotechnology Journal, 13(3), 409–420. https://doi.org/10.1111/pbi.12346
Senthil, K., Jayakodi, M., Thirugnanasambantham, P., Lee, S. C., Duraisamy, P., Purushotham, P. M., Rajasekaran, K., Charles, S. N., Roy, I. M., Nagappan, A. K., Kim, G. S., Lee, Y. S., Natesan, S., Min, T. S., & Yang, T. J. (2015). Transcriptome analysis reveals in vitro cultured Withania somnifera leaf and root tissues as a promising source for targeted withanolide biosynthesis. BMC Genomics, 16, 1–16.
Patel, R. K., & Jain, M. (2012). NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One, 7(2), e30619. https://doi.org/10.1371/journal.pone.0030619
Kozomara, A., & Griffiths-Jones, S. (2014). miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Research, 42(D1), D68–D73. https://doi.org/10.1093/nar/gkt1181
Numnark, S., Mhuantong, W., Ingsriswang, S., & Wichadakul, D. (2012). C-mii: a tool for plant miRNA and target identification. BMC Genomics, 13(Suppl 7), S16. https://doi.org/10.1186/1471-2164-13-S7-S16
Conesa, A., Gotz, S., García-Gómez, J.M., Terol, J., Talón, M. & Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21(18), 3674–3676
Botton, A., Galla, G., Conesa, A., Bachem, C., Ramina, A. & Barcaccia, G. (2008). Large-scale Gene Ontology analysis of plant transcriptome-derived sequences retrieved by AFLP technology. BMC Genomics, 9(1), 347
Galla, G., Barcaccia, G., Ramina, A., Collani, S., Alagna, F., Baldoni, L., Cultrera, N.G., Martinelli, F., Sebastiani, L. & Tonutti, P., (2009). Computational annotation of genes differentially expressed along olive fruit development. BMC Plant Biology, 9(1), 128
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., Amin, N., Schwikowski, B., & Ideker, T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. https://doi.org/10.1101/gr.1239303
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., & Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Research, 35(Web Server), W182–W185. https://doi.org/10.1093/nar/gkm321
Singh, R., Pandey, N., Naskar, J., & Shirke, P. A. (2015). Physiological performance and differential expression profiling of genes associated with drought tolerance in contrasting varieties of two Gossypium species. Protoplasma, 252(2), 423–438. https://doi.org/10.1007/s00709-014-0686-0
Singh, P., Guleri, R., Angurala, A., Kaur, K., Kaur, K., Kaul, S. C., Wadhwa, R., & Pati, P. K. (2017). Addressing challenges to enhance the bioactives of Withania somnifera through organ, tissue, and cell culture based approaches. BioMed Research International, 2017, 1–15. https://doi.org/10.1155/2017/3278494
Dhar N, Razdan S, Rana S, Bhat WW, Vishwakarma R, Lattoo SK (2015) A decade of molecular understanding of withanolide biosynthesis and in vitro studies in Withania somnifera (L.) Dunal: prospects and perspectives for pathway engineering. Frontiers in Plant Science 6. doi:https://doi.org/10.3389/fpls.2015.01031.
Pal, S., Yadav, A. K., Singh, A. K., Rastogi, S., Gupta, M. M., Verma, R. K., Nagegowda, D. A., Pal, A., & Shasany, A. K. (2017). Nitrogen treatment enhances sterols and withaferin A through transcriptional activation of jasmonate pathway, WRKY transcription factors, and biosynthesis genes in Withania somnifera (L.) Dunal. Protoplasma, 254(1), 389–399. https://doi.org/10.1007/s00709-016-0959-x
Davis, L., & Kuttan, G. (2000). Immunomodulatory activity of Withania somnifera. Journal of Ethnopharmacology, 71(1-2), 193–200. https://doi.org/10.1016/S0378-8741(99)00206-8
Khanna, D., Sethi, G., Ahn, K. S., et al. (2007). Natural products as a gold mine for arthritis treatment. CurrOpinPharmacol, 7, 344–351.
Ku, S. K., & Bae, J. S. (2014). Antiplatelet, anticoagulant, and profibrinolytic activities of withaferin A. VascPharmacol, 60, 120–126.
Datta, A., Bagchi, C., & Tripathi, S. K. (2013). Antidiabetic and antihyperlipidemic activity of hydroalcoholic extract of Withania coagulans Dunal dried fruit in experimental rat models. J Ayurveda Integr Med, 4, 99–106.
Choudhary, S., Kumar, P., & Malik, J. (2013). Plants and phytochemicals for Huntington’s disease. Pharmacognosy Reviews, 7(14), 81–91. https://doi.org/10.4103/0973-7847.120505
Pingali, U., Pilli, R., & Fatima, N. (2014). Effect of standardized aqueous extract of Withania somnifera on tests of cognitive and psychomotor performance in healthy human participants. Pharmacogn Res, 6(1), 12–18. https://doi.org/10.4103/0974-8490.122912
Rai, M., Jogee, P. S., Agarkar, G., & dos Santos, C. A. (2016). Anticancer activities of Withania somnifera: current research, formulations, and future perspectives. Pharmaceutical Biology, 54(2), 189–197. https://doi.org/10.3109/13880209.2015.1027778
Griffiths, J. S., Saini, H. K., Van, D. S., & Enright, A. J. (2008). miRBase: tools for microRNA genomics, Nucleic Acids Res., 36, D154eD158.
Xu, Y., Chu, L., Jin, Q., Wang, Y., Chen, X., Zhao, H., & Xue, Z. (2015). Transcriptome-wide identification of miRNAs and their targets from Typhaangustifolia by RNA-Seq and their response to cadmium stress. PLoS One, 10(4), e0125462. https://doi.org/10.1371/journal.pone.0125462
Saifi, M., Nasrullah, N., Ahmad, M. M., Ali, A., Khan, J. A., & Abdin, M. Z. (2015). In silico analysis and expression profiling of miRNAs targeting genes of steviol glycosides biosynthetic pathway and their relationship with steviol glycosides content in different tissues of Stevia rebaudiana. Plant Physiology and Biochemistry, 94, 57–64. https://doi.org/10.1016/j.plaphy.2015.05.009
Koul, A., Yogindran, S., Sharma, D., Kaul, S., Rajam, M. V., & Dhar, M. K. (2016). Carotenoid profiling, in silico analysis and transcript profiling of miRNAs targeting carotenoid biosynthetic pathway genes in different developmental tissues of tomato. Plant Physiology and Biochemistry, 108, 412–421. https://doi.org/10.1016/j.plaphy.2016.08.001
Sharma, D., Tiwari, M., Pandey, A., Bhatia, C., Sharma, A., & Trivedi, P. K. (2016). MicroRNA858 is a potential regulator of phenylpropanoid pathway and plant development in Arabidopsis. Plant Physiology, pp.01831.2015. https://doi.org/10.1104/pp.15.01831
Gupta, O. P., Karkute, S. G., Banerjee, S., Meena, N. L., & Dahuja, A. (2017). Contemporary understanding of miRNA-based regulation of secondary metabolites biosynthesis in plants. Frontiers in Plant Science, 8. https://doi.org/10.3389/fpls.2017.00374
Bologna, N. G., Mateos, J. L., Bresso, E. G., & Palatnik, J. F. (2009). A loop-to-base processing mechanism underlies the biogenesis of plant microRNAs miR319 and miR159. The EMBO Journal, 28(23), 3646–3656. https://doi.org/10.1038/emboj.2009.292
Bologna, N. G., Schapire, A. L., Zhai, J., Chorostecki, U., Boisbouvier, J., Meyers, B. C., & Palatnik, J. F. (2013). Multiple RNA recognition patterns during microRNA biogenesis in plants. Genome Research, 23(10), 1675–1689. https://doi.org/10.1101/gr.153387.112
Das, A., Das, P., Kalita, M. C., & Mondal, T. K. (2016). Computational identification, target prediction, and validation of conserved miRNAs in insulin plant (Costuspictus D. Don). Applied Biochemistry and Biotechnology, 178(3), 513–526. https://doi.org/10.1007/s12010-015-1891-9
Xu, S., Jiang, Y., Wang, N., Xia, B., Jiang, Y., Li, X., Zhang, Z., Li, Y., & Wang, R. (2016). Identification and differential regulation of microRNAs in response to methyl jasmonatetreatment in Lycorisaurea by deep sequencing. BMC Genomics, 17(1), 789. https://doi.org/10.1186/s12864-016-2645-y
Xu, W., Cui, Q., Li, F., & Liu, A. (2013). Transcriptome-wide identification and characterization of microRNAs from castor bean (Ricinuscommunis L.) PLoS One, 8(7), e69995. https://doi.org/10.1371/journal.pone.0069995
Zhang, B. H., Pan, X. P., Cox, S. B., Cobb, G. P., & Anderson, T. A. (2006). Evidence that miRNAs are different from other RNAs. Cellular and Molecular Life Sciences CMLS, 63(2), 246–254. https://doi.org/10.1007/s00018-005-5467-7
Yang, Y., Chen, X., Chen, J., Xu, H., Li, J., & Zhang, Z. (2011). Differential miRNA expression in Rehmanniaglutinosa plants subjected to continuous cropping. BMC Plant Biology, 11(1), 53. https://doi.org/10.1186/1471-2229-11-53
Kaur, P., Shukla, N., Joshi, G., VijayaKumar, C., Jagannath, A., Agarwal, M., Goel, S., & Kumar, A. (2017). Genome-wide identification and characterization of miRNAome from tomato (Solanum lycopersicum) roots and root-knot nematode (Meloidogyne incognita) during susceptible interaction. PLoS One, 12(4), e0175178. https://doi.org/10.1371/journal.pone.0175178
Lu, Y.-B., Qi, Y.-P., Yang, L.-T., Guo, P., Li, Y., & Chen, L.-S. (2015). Boron-deficiency-responsive microRNAs and their targets in Citrus sinensis leaves. BMC Plant Biology, 15(1). https://doi.org/10.1186/s12870-015-0642-y
Dutt, S., Manjul, A. S., Raigond, P., Singh, B., Siddappa, S., Bhardwaj, V., Kawar, P. G., Patil, V. U., & Kardile, H. B. (2017). Key players associated with tuberization in potato: potential candidates for genetic engineering. Critical Reviews in Biotechnology, 1–19(7), 942–957. https://doi.org/10.1080/07388551.2016.1274876
Miozzi, L., Napoli, C., Sardo, L., & Accotto, G. P. (2014). Transcriptomics of the interaction between the monopartite phloem-limited geminivirus tomato yellow leaf curl Sardinia virus and Solanum lycopersicum highlights a role for plant hormones, autophagy and plant immune system fine tuning during infection. PLoS One, 9(2), e89951. https://doi.org/10.1371/journal.pone.0089951
Khaldun, A. B. M., Huang, W., Liao, S., Lv, H., & Wang, Y. (2015). Identification of MicroRNAs and target genes in the fruit and shoot tip of Lyciumchinense: a traditional Chinese medicinal plant. PLoS One, 10(1), e0116334. https://doi.org/10.1371/journal.pone.0116334
Gao, C., Wang, P., Zhao, S., Zhao, C., Xia, H., Hou, L., Ju, Z., Zhang, Y., Li, C., & Wang, X. (2017). Small RNA profiling and degradome analysis reveal regulation of microRNA in peanut embryogenesis and early pod development. BMC Genomics, 18(1), 220. https://doi.org/10.1186/s12864-017-3587-8
Song, C., Yu, M., Han, J., Wang, C., Liu, H., Zhang, Y., & Fang, J. (2012). Validation and characterization of Citrus sinensis microRNAs and their target genes. BMC Research Notes, 5(1), 235. https://doi.org/10.1186/1756-0500-5-235
Omidvar, V., Mohorianu, I., Dalmay, T., & Fellner, M. (2015). Identification of miRNAs with potential roles in regulation of anther development and male-sterility in 7B-1 male-sterile tomato mutant. BMC Genomics, 16(1). https://doi.org/10.1186/s12864-015-2077-0
Liang, G., Li, Y., He, H., Wang, F., & Yu, D. (2013). Identification of miRNAs and miRNA-mediated regulatory pathways in Carica papaya. Planta, 238(4), 739–752. https://doi.org/10.1007/s00425-013-1929-6
Mangrauthia, S. K., Bhogireddy, S., Agarwal, S., Prasanth, V. V., Voleti, S. R., Neelamraju, S., & Subrahmanyam, D. (2017). Genome-wide changes in microRNA expression during short and prolonged heat stress and recovery in contrasting rice cultivars. Journal of Experimental Botany, 68(9), 2399–2412. https://doi.org/10.1093/jxb/erx111
Campbell, J. A., Davies, G. J., Bulone, V., & Henrissat, B. (1997). A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. The Biochemical Journal, 326(3), 929–939. https://doi.org/10.1042/bj3260929
Jones, P., & Vogt, T. (2001). Glycosyltransferases in secondary plant metabolism: tranquilizers and stimulant controllers. Planta, 213(2), 164–174. https://doi.org/10.1007/s004250000492
Bhat, W. W., Lattoo, S. K., Razdan, S., Dhar, N., Rana, S., Dhar, R. S., Khan, S., & Vishwakarma, R. A. (2012). Molecular cloning, bacterial expression and promoter analysis of squalene synthase from Withania somnifera (L.) Dunal. Gene, 499(1), 25–36. https://doi.org/10.1016/j.gene.2012.03.004
Chen, X., Dai, G., Ren, Z., Tong, Y., Yang, F., & Zhu, Y. (2016). Identification of dietetically absorbed rapeseed (Brassica campestris L.) bee pollen microRNAs in serum of mice. BioMed Research International, 2016, 1–5. https://doi.org/10.1155/2016/5413849
Chin, A. R., Fong, M. Y., Somlo, G., Wu, J., Swiderski, P., Wu, X., & Wang, S. E. (2016). Cross-kingdom inhibition of breast cancer growth by plant miR159. Cell Research, 26(2), 217–228. https://doi.org/10.1038/cr.2016.13
Liang, G., Zhu, Y., Sun, B., Shao, Y., Jing, A., Wang, J., & Xiao, Z. (2014). Assessing the survival of exogenous plant microRNA in mice. Food Science & Nutrition, 2(4), 380–388. https://doi.org/10.1002/fsn3.113
Acknowledgements
We would like to thank SRA database of NCBI database for making it freely available to the scientific community. Financial assistance to SS under BTISnet programme of DBT-New Delhi, S and RS under SERB-PDF and GS under ICMR-SRF is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Figure 1
(DOCX 892 kb)
Supplementary Figure 2
(DOCX 583 kb)
Supplementary Figure 3
(DOCX 507 kb)
Supplementary Figure 4
(DOCX 502 kb)
Supplementary Table 1
(DOCX 16 kb)
Supplementary Table 2
(DOCX 36 kb)
Supplementary Table 3
(DOCX 15 kb)
Supplementary Table 4
(DOCX 30 kb)
Supplementary Table 5
(DOCX 58 kb)
Rights and permissions
About this article

Cite this article
Srivastava, S., Sanchita, Singh, R. et al. Comparative Study of Withanolide Biosynthesis-Related miRNAs in Root and Leaf Tissues of Withania somnifera. Appl Biochem Biotechnol 185, 1145–1159 (2018). https://doi.org/10.1007/s12010-018-2702-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-018-2702-x