
Abstract
B cells and in particular antibodies has always played second fiddle to cellular immunity in regard to tuberculosis (TB). However, recent studies has helped position humoral immunity especially antibodies back into the foray in relation to TB immunity. Therefore, the ability to correlate the natural antibody responses of infected individuals toward TB antigens would help strengthen this concept. Phage display is an intriguing approach that can be utilized to study antibody-mediated responses against a particular infection via harvesting the B cell repertoire from infected individuals. The development of disease-specific antibody libraries or immune libraries is useful to better understand antibody-mediated immune responses against specific disease antigens. This study describes the generation of an immune single-chain variable fragment (scFv) library derived from TB-infected individuals. The immune library with an estimated diversity of 109 independent clones was then applied for the identification of monoclonal antibodies against Mycobacterium tuberculosis α-crystalline as a model antigen. Biopanning of the library isolated three monoclonal antibodies with unique gene usage. This strengthens the role of antibodies in TB immunity in addition to the role played by cellular immunity. The developed library can be applied against other TB antigens and aid antibody-derived TB immunity studies in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tuberculosis (TB) is one of the leading causes of infectious disease-related mortality worldwide [1]. It is prominent intracellular pathogen for human. The causative agent for TB is Mycobacterium tuberculosis (Mtb), a gram-positive acid-fast bacterium. Treatment of TB takes up to several months due to the slow-growing nature of this intracellular organism [2]. The conventional treatment for TB infections in humans requires the use of appropriate antibiotics. The treatment regime normally involves the use of an antibiotic cocktail given over a period of 6 months and may stretch in some cases to a year. However, World Health Organization (WHO) reported that many Mtb strains have shown the ability to develop resistance to one or more of the standard TB drugs, which complicates treatment greatly [3]. Therefore, a new suitable alternative treatment for TB is highly required.
TB immunity has longed been associated with the role of cellular immunity mainly due to the notion that the humoral immunity does not contribute to the protection against intracellular pathogens and is mainly attributed only to cellular defense mechanisms. This matter is further complicated with the variation in antibody responses accorded to different individuals. However, the protection afforded by antibodies is now seen for a range of intracellular pathogens via Fc-receptor phagocytosis. An in-depth review on the role antibodies play in TB immunity can be read in [4]. However, the ability to harness antibodies for the treatment of TB is something intriguing that has attracted much attention. A very important notion in harnessing antibodies for therapy of TB in humans is the ability to isolate and identify quality antibodies against specific antigens.
Phage display is currently the gold standard for the identification and isolation of human monoclonal antibody (mAb) in vitro using a series of selection and enrichment steps from a library of antibodies [5]. The physical presentation of antibodies on phage particles is accomplished by the incorporation of the DNA encoding an antibody as a fusion to the coat protein III (pIII) gene. Therefore, by collecting antibody genes from individuals, a collection of the natural antibody repertoire of a person can be replicated in vitro in the form of a phage library. Among the three main antibody phage display libraries (immune, naïve, and synthetic antibody libraries), immune libraries are most interesting for disease-associated applications. This is mainly related to the source for the library generation, where immune libraries focus on the use of materials from infected patients [5]. The logic is to apply the skewed immune response by infected individuals to provide the repertoire of antibodies against disease-specific proteins [5]. This allows for a physical representation of the antibody responses during infections to be studied or applied for mAb generation. The application of immune libraries to isolate disease-specific mAbs has been successfully applied for several diseases over the years [6, 7]. Antibodies produced using conventional immunization like that in mice may target similar antigens as antibodies produced using human antibody libraries, but the side effects are a cause of concern [8]. Although targeting similar targets, the variation between the species may render such antibodies useless against TB infections. Therefore, the use of human antibodies for downstream applications is of paramount importance. However, the main challenges associated with the application of immune libraries are the availability of sample material for library generation and the need to generate new libraries for different diseases.
Phage display allows for a simple laboratory scale assessment on antibody-based immunity against TB. We sought to apply an immune TB antibody library derived from infected individuals to screen for potential antibodies against a known TB antigen. α-crystalline or HSP16.3 is a small heat shock protein of Mtb that is expressed during the stationary phase [9]. α-crystalline is a membrane protein responsible for Mtb cell wall thickening and found to illicit antibody responses specific for the M. tuberculosis complex [10]. It is a dominant protein produced by Mtb during the latent stage of infection but not during exponential phase of infection [11]. The α-crystalline antigen is present on the surface of tubercle bacilli and is highly expressed in organisms growing within infected macrophages. There was also a report of mAbs-specific toward α-crystalline, arabinomannan, and liporabinomannan that conferred protection in mice [12]. It has also been reported that a chicken single-chain variable fragment (scFv) antibody library was used to generate monoclonal antibodies against the α-crystalline antigen [13]. This is a good starting point for the potential application of mAbs against these antigens for therapeutic application in humans. Although it is possible that the immune response against these proteins conferred by mice may differ in humans, nevertheless, the generation of human antibodies against these proteins would be critical to validate its use in humans. This makes α-crystalline a suitable model antigen to investigate if the natural antibody repertoire of TB-infected individuals can indeed mediate antibody-based responses against TB antigens.
Here, we describe the generation and application of a TB immune antibody library generated with samples derived from TB-infected individuals. The generated library will function to resemble the natural B cell repertoire post TB infection. We hypothesize that the availability of a natural B cell immunity toward TB will help prime the antibody responses to generate a preferential repertoire of antibodies against TB-associated antigens. In order to validate the library, selection using α-crystalline as a model target antigen for monoclonal antibody enrichment was carried out. Successful isolation of mAbs against the model target antigen will help to strengthen the concept of antibody-based immunity against TB and utilization of antibody phage display as a tool to represent the in vivo immune system by in vitro methods. The work also highlights the prospect of immune antibody libraries as a valuable system for infectious diseases in general. More importantly, the antibody library generated can be applied to continuously generate monoclonal antibodies against other TB target antigens that can be beneficial for the diagnosis and therapy of TB.
Materials and Methods
Construction of Immune Library
Blood samples from six TB-infected individuals were collected in accordance with the protocols approved by the human research ethics of Universiti Sains Malaysia Human Ethics Committee. All donors aged between 18 and 61 years old were confirmed positive with active TB by sputum smear, culture, and x-ray. The blood specimens were collected using EDTA blood collection tubes for total lymphocyte isolation. Sample processing was carried out according to previously published protocol [7].
A set of V-gene-specific primers was used to amplify the antibody variable gene repertoire [14]. The primers were synthesized by First BASE Laboratories Sdn. Bhd, Malaysia and Integrated DNA Technologies Pte. Ltd. (IDT), Singapore. The polymerase chain reaction (PCR) amplification and purification was optimized for both the light chain (VL) and heavy chain (VH) according to previously published protocol with different types of polymerases [7, 15]. A 20-μL PCR reaction was carried out with 2 μL of 10× DreamTaq buffer and 0.2 μL (2 U) DreamTaq DNA with initial denaturation at 95 °C for 90 s, followed by 30 cycles at 95 °C for 30 s, annealing at 55 °C for 30 s, elongation at 72 °C for 1 minute (min), and the final extension step involved incubation for 5 min at 72 °C. The second PCR amplification was carried out in similar fashion to introduce the restriction enzyme (RE) cut sites to the genes for cloning. A total of 7 VH genes, 9 Vλ genes, and 5 Vκ genes were individually amplified and pulled based on their V-gene family prior to separation by 1% agarose gel electrophoresis.
The scFv TB library cloning was carried out using a two-step cloning procedure to clone the VH and VL and repertoire into pLABEL, a phagemid vector. The digestion and ligation was performed according to previously published protocol with slight modifications [7, 15]. The amplified VH repertoire and pLABEL phagemid were digested with XhoI and NcoI RE (New England Biolabs) and subsequently cloned into pLABEL to generate a mini VH library in DH10B Escherichia coli (E.coli) cells (Agilent Technologies, Santa Clara, CA, USA). Five hundred nanograms of DNA template was used with 20 U of XhoI and NcoI RE for every 50 μL digestion. The enzyme was heat inactivated at 70 °C for 20 min. The digested products were separated by agarose gel electrophoresis and purified using QiaQuick Gel extraction kit (Qiagen) according to the manufacturer’s protocol and eluted with 40 μL of elution buffer. The purified digested products of the VH repertoire and pLABEL were ligated together to generate a mini VH library. A 1:5 vector to insert ratio with 300 ng of pLABEL was used for every ligation reaction with 3 U of T4 DNA ligase (New England Biolabs). The ligation and transformation reaction was also carried out according to previously published protocol [7, 15]. This process was continued until a total of six dense agar plates (25 cm2) were obtained. The mini VH library was stored at −80 °C in 20% glycerol.
For the second step of cloning, the same protocol was applied to complete the full immune scFv TB library. The purified VL repertoire and mini VH library were digested with SalI and NotI RE (New England Biolabs). The PCR product was adjusted to reach a 1:7 vector to insert ratio using 300 ng mini VH library for ligation as described earlier. The ligated product was transformed using electrocompetent XL1 Blue E. coli cells (Agilent Technologies, Santa Clara, CA, USA). This process was continued until a total of six dense agar plates (25 cm2) were obtained. The library size was estimated by taking 10 μL of the pooled culture from six agar plates. A tenfold serial dilution was carried out with 2-YT broth until 10−12 and was plated out on 2-YT agar plate. Library cloning efficiency was determined by colony PCR and DNA sequencing of random colonies.
Antigen Preparation
The expression vector with the α-crystalline gene was transformed into BL21 (DE3) E. coli strain (Invitrogen, Carlsbad, CA, USA) and expression was carried out according to the previously described protocol [16]. The pellet from the harvested cells was re-suspended in ice-cold lysis buffer. Lysozyme (0.5 mg/ml) was added to the mixture followed by the addition of protease inhibitors. The mixture was then incubated on ice for 30 min and then subjected to cell breakage. The antigen was then purified using a 1-mL Ni-NTA Agarose fast flow column (GE healthcare, Sweden) according to the manufacturer’s instructions. The purified biotinylated α-crystalline antigen fractions were analyzed by 12% SDS-PAGE and Western blot.
Biopanning of Immune Human scFv Antibody Library
Biopaninng of the immune scFv TB library with the purified α-crystalline antigen was carried out using the standard microtitre plate panning protocol according to previously published protocol with slight modifications [7, 15]. The wells were pre-coated with 10 μg/well of α-crystalline target antigen at 4 °C, overnight. Next day, the wells were blocked with PBST-M (3% skim milk in PBS and 0.1% v/v Tween 20) (Nacalai Tesque, Inc., Nijo Karasuma, Kyoto, Japan) for 1 hour (h) at room temperature (rt). The bound antibody phages were eluted with 100 μL trypsin (10 μg/mL) solution.
Polyclonal ELISA
Polyclonal phage ELISA was performed as previously published protocol [7, 14, 15]. The wells were coated with 10 μg/well α-crystalline antigen at 4 °C overnight (o/n). Positive and negative controls were also included in the ELISA assay. The positive control well was coated with eGFP protein, while the negative control well was left uncoated. All coated wells were blocked with 3% skim milk in PBST.
Monoclonal ELISA
Monoclonal phage ELISA was performed with standard protocol according to previously published protocol [7, 15] with three replicates. Monoclonal phage antibody ELISA was performed with clones from round 2 and round 3 of the panning process. A total of 184 colonies were picked and screened. Two wells were left for positive (eGFP protein) and negative (empty) control. The plates containing single colonies were used for ELISA.
DNA Sequencing
DNA sequencing of the clones were carried out using previously published primers by FirstBase Sdn Bhd [7, 15]. DNA sequencing data was analyzed using ContigExpress from Vector NTI 11 (Invitrogen). V-gene identification and complementarity-determining regions (CDR) composition analysis was carried out using IMGT/V-QUEST [17, 18]. These tools also provide the CDR sequences and length analysis.
Soluble Antibody Production
The antibody clones obtained earlier were transformed into BL21 (DE3) E. coli strain. The culture was then plated on 2-YT plate with 100 μg/ml ampicillin and 2% glucose at 37 °C for overnight. The next day a single colony was picked, and the protein expression was carried out. Soluble antibody was carried out using the previously published protocol modified with OD600 ~ 0.5 IPTG induction at 25 °C with 160 rpm shakingo/n [15]. The culture was harvested by centrifugation at 10000×g for 30 min at 4 °C, and the pellet was kept at − 20 °C until use. The antibody clones were then purified using 1 mL Ni-NTA Agarose fast flow column (GE healthcare, Sweden) according to the manufacturer’s instructions. The purified fractions of the clones were analyzed by 12% SDS-PAGE and Western blot.
Soluble Antibody ELISA
The binding of soluble antibodies with the α-crystalline target antigen was confirmed by ELISA assay. A total of 50 μg/well of purified antibody was immobilized on the well o/n. The wells were washed 3× with PBST and blocked for 1 h with 150 μL 3% milk powder in PBST (PTM) (Nacalai Tesque, Inc., Nijo Karasuma, Kyoto, Japan). After 1 h, the wells were washed again before 100 μL (10 μg/mL) of target biotinylated antigen was added to the well. The wells were incubated for 1 h for the binding of antibody with target antigen. The wells were washed 3× with PBST and 100 μL of streptavidin-HRP in 3% PTM (1:2500) was added. After that, 100 μL 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid (ABTS) development buffer was added to each well and incubated in the dark for 30 mins before readings were taken using a SkanlT absorbance reader (Thermo Scientific, USA) at 405 nm. For background control, wells were blocked with PTM without the presence of antibodies. Then 10 μg/well of biotinylated antigen was added to the wells in PBS buffer and detected with streptavidin-HRP.
Titration ELISA
The titration ELISA was performed in a similar manner as the soluble antibody ELISA but with slight modifications. Different concentrations of purified monoclonal antibodies ranging from 10 pg/well to 1 mg/well were immobilized on the well o/n. The wells were then washed 3× with PBST and blocked with PTM for 1 h. The wells were then washed 3× with PBST. A total of 100 μL (10 μg/mL) of target biotinylated antigen was added to each well and incubated for 1 h for binding. The wells were washed 3× with PBST and 100 μL of streptavidin-HRP in 3% PTM (1:2500) was added. After that, 100 μL ABTS development buffer was added to each well and incubated in the dark for 30 mins before readings were taken using a SkanlT absorbance reader (Thermo Scientific, USA) at 405 nm.
Competitive ELISA
Competitive ELISA was used to determine the binding site of the monoclonal antibodies for three different clones monoclonal antibody. Competitive ELISA was performed by coating the wells with 10 μg/well α-crystalline antigen 4 °C o/n. Then, wells were washed with PBST and incubated with 100 μL of monoclonal antibody protein followed by incubation with monoclonal antibody phage. Both incubations were performed for 1 h. Wells were then washed and incubated with 100 μL HRP conjugated anti-M13 antibody 1:5000 diluted in PTM for 1 h at rt with shaking. Plate was washed, and ABTS substrate was added, and the absorbance was read at 405 nm using a SkanlT absorbance reader (Thermo Scientific, USA).
Results
Library Cloning
The immune scFv TB library was generated for the isolation of monoclonal scFv fragments to α-crystalline antigen using two-step cloning. The first cloning step generated a mini VH antibody library with an estimated library size of 107. This was followed up with the second cloning step involving the introduction of the VL repertoire to the mini VH library to yield a final scFv TB immune antibody library with an estimated diversity of 109. Fifteen single colonies were randomly selected for colony PCR using LMB3 forward primer, and PIII reverse primer to yield an approximate band size of ~ 1100 bp indicating both VL and VH were successfully cloned into pLABEL (data not shown). The cloning efficiency was estimated to be 60%. Sequencing of 10 random clones showed that 50% of the clones had sequences without any mutation or frame shift (Table 1). The remaining clones presented truncated sequences or frame shift due to indels.
Antigen Preparation
The α-crystalline antigen was expressed, purified, and analyzed with 12% SDS-PAGE to yield an expected band at ~ 20 kDa (Fig. 1a). α-crystalline was successfully expressed as shown in the SDS gel, and the purified product showed good yield with acceptable purity. Western blot analysis result showed that the biotinylated antigen could be detected using streptavidin-HRP with the correct band size (Fig. 1b). The purified fraction was then used for panning experiments.

Analysis of target antigen expression and purification. a SDS-PAGE profile of purified α-crystalline recombinant antigen. M Bluelf pre-stained protein ladder, L1 pellet, L2 crude, L3 flow-through, L4 last wash, L5 elution1, L6 elution 2, L7 elution 3, L8 elution 4. b Western Blot analysis of α-crystalline antigen. M Bluelf pre-stained ladder, L1 -ve, L2 positive control, L3 α-crystalline antigen
Biopanning and Polyclonal ELISA
Three rounds of biopanning using the immune scFv TB library against α-crystalline antigen were carried out using the conventional method plate panning protocol. The immune scFv TB library against α-crystalline antigen showed gradual enrichment from round one to round three, with the highest enrichment in round three (Table 2).
The amount of starting phage particles for each round was 8 × 1010, 4.70 × 1010, and 7 × 109 pfu for rounds 1 to 3, respectively. The amount of recovered phage particles for the three successive rounds was 9 × 105, 10 × 104, and 5 × 106 pfu. The enrichment ratio takes into account the amount of phage recovered over the amount of phage used. The enrichment ratio for all three rounds was 1.1 × 10−5, 2.5 × 10−6, and 7 × 10−4, respectively. The enrichment ratio from rounds 2 to 3 in comparison to round 1 was 22.72 and 63.6-folds, respectively. The enrichment pattern correlates with the increasing trend of the polyclonal ELISA results with OD405 at 0.4, 0.9, and 1.5 for the three rounds of biopanning (Fig. 2).

Polyclonal enrichment of scFv from each panning round by ELISA
Monoclonal ELISA Control
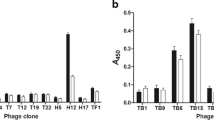
A total of 184 antibody clones were randomly picked from rounds 2 and 3 of the panning process. The result of 26 monoclonal clones indicated the positive binders with high absorbance (Fig. 3) compared to background value. The suffix P1 indicates the plate number, and E1 identifies the well position in the plate. The positive clones were identified with an OD405 value above 0.1 after deducting the background values. The library generated was able to provide satisfactory enrichment of α-crystalline-specific scFv TB clones. The value of the negative control (H3, H6, H9, and H12) showed an OD405 value of 0.1; therefore, all the binders reading below OD405 value of 0.1 were not taken into consideration.

Monoclonal phage ELISA of randomly selected scFv tested against α-crystalline
All positive monoclonal antibody clones were analyzed by colony PCR to evaluate the insert before sequencing. From 26 positive clones, only 10 clones showed the correct band size, which was approximately around 1100 bp (data not shown). The identified clones were then sent for DNA sequencing and analyzed.
Antibody Gene Analysis
Ten antibody clones were analyzed by Vbase2 software based on the human germline sequence. There were showed only three antibody clones that have full sequence of VH, and VL fragments without any frameshift mutations were used for expression and downstream analysis. The remaining seven clones showed either truncation or frame shift. The gene segment usage of the three clones was shown in Table 3. The antibody sequence was analyzing using VBASE2 software based on the human germline sequence [19]. The clones were IGKV3 and IGHV2 for clone 1H, IGKV4 and IGHV2 for clone 1B, and IGLV1 and IGHV1 for clone 10G.
The CDR length of the variable domains is an important characteristic for the binding site topology of antibody clones especially that of the CDR3 in the heavy chain [7]. The analysis of both VH and VL repertoires showed that clones 1H and 1B presented the same J segment IGHJ3*02 and IGKJ4*01. However, both clones showed diversification in the D segment usage with gene segment D310*01 and D310*02 being preferred.
The VH CDR1 exhibited a length of 10 amino acids for clones 1H and 1B. However, clone 10G presented a shorted CDR1 with 8 amino acids. The lengths of CDR2 were different across all three clones with 6, 7, and 8 amino acids for 1B, 1H, and 10G, respectively. The CDR3 length distribution for clones 1H, 1B and 10G was 22, 24 and 13 amino acids. Human CDR3 length was extremely broad and normally distributed with an average between 1 and 35 aa distribution resembling a Gaussian distribution with an average loop length of 15.2 (± 4.1) [20]. A longer D gene segment results in CDR3 with longer lengths, which could be due to the gene conversion process generating extended N regions [21]. The VL repertoire showed 7, 8, and 12 aa (clones 1H, 10G, and 1B) for CDR1 with a similar length for CDR2 in all clones with 3 amino acids. The VL CDR3 length distribution ranged from 9 to 11 amino acid residues, respectively.
Analysis of Amino Acid Distribution
Figure 4 shows the overall amino acid propensities for the VH and VL domains for the enriched clones. The CDR1 of the VH domain shows a higher representation of serine followed by glycine. Serine is useful for antigen recognition [7]. However, in the CDR2 of the VH domains, a higher representation for aspartic acid and arginine was recorded. The CDR3 of the VH showed a higher representation in phenylalanine in comparison to other aa. The VL domain also showed a higher distribution of serine in CDR1. The distribution of the VL CDR2 had higher distributions of serine and asparagine, while serine and threonine was abundant for CDR3. This analysis shows that CDR2 and CDR3 for VH and VL are prone to exhibit small-sized amino acids and will most likely contribute to the antigen-binding ability of antibody clones [7]. Human CDR had lower cysteine distribution around 1.6% but had higher tyrosine around 16.8% [22].

Amino acid frequency analysis of CDR1, CDR2, and CDR3 amino acid distribution of isolated anti-α-crystalline scFv monoclonal antibodies. Color keys represent the different amino acids
Soluble scFv Expression and Purification
The three clones that contained the full antibody sequence were expressed and purified. The SDS analysis shows that the clones were successfully but with moderate yields (Fig. 5). The binding characteristic of the three clones was further confirmed by Western blot with all three clones at 35 kDa binding to the target antigen. The results showed that even with a moderate expression yields, the antibody clones were still capable of producing sufficient soluble scFv for detection with the target antigen using Western blot. This shows the ability of the soluble antibody clones to maintain its binding functionality against the target antigen even in reducing conditions.

Expression and purification analysis of soluble anti-α-crystalline scFv monoclonal antibodies. a, b, and c shows the SDS-PAGE analysis of soluble monoclonal antibody clones purified using his-tag column. A clone 1H, B clone 1B, C clone 10G, M Bluelf pre-stained protein ladder, L1 pellet, L2 supernatant, L3 flow-through, L4 wash 1, L5 elution1, L6 elution 2. d Western blot detection of soluble antibody clones against α-crystalline antigen. Western blot analysis was carried out where M: Bluelf pre-stained ladder; L1 positive control (ubiquitin protein); L2 1B, L3 1H, L4 10G
Soluble scFv Antibody Protein ELISA Against α-Crystalline Antigen
Soluble antibody ELISA was performed to confirm the binding of the soluble scFv antibodies 1H, 10G, and 1B clone against α-crystalline antigen. IgG 1H, 10G, and 1B α-crystalline antigen monoclonal antibody showed a positive signal and maintained its functionality in soluble form with OD405 after normalizing with background (Fig. 6a). Figure 6b shows the titration ELISA results of the scFv clones with the continuous line indicating the signal (absorption at 405 nm) resulting from the binding of the scFv antibody against α-crystalline antigen at different concentrations. The OD readouts for the clones at pg levels were low with a gradual increase in OD readouts with an increasing amount of antigen. The signal increment of all three clones shows a direct correlation to the amount of antibody used at each concentration. The overall pattern indicates that clone 1B exhibited stronger binding characteristics compared to the other two clones, 10G and 1H.

Analysis of binding characteristics of enriched scFv monoclonal antibodies against α-crystalline. a ELISA result of enriched monoclonal scFv clones against α-crystalline antigen. b Titration-based ELISA for clone 10G, 1H, and 1B against α-crystalline antigen
Competitive ELISA
The absorbance reading of the competitive ELISA will decrease when two different clones are competing for the same binding site and vice versa. Absorbance reading above OD 0.5 was determined as not competing. The three monoclonal antibodies were competed against each other to determine the competitive binding nature of the clones. All three monoclonal antibodies were represented by unique V-gene family sequences. This means that all three clones were independently unique in sequence. The analysis shows that clone 1H with IgHV2-VK3 combination competed with clone 1B that has a IgHV2-VK4 combination. However, clone 10G with the IgHV1-LV1 gene combination did not show any competition with the other two clones (Fig. 7).

Competitive ELISA of enriched scFv monoclonal antibody protein with monoclonal antibody phage against α-crystalline. As a control, each antibody protein was incubated without a competitor
Discussion
Cellular immunity for TB infection has been well documented, but the inherent role of the humoral immunity, specifically antibody responses, has been overshadowed by T cell responses. Taking a closer look at the immunological mechanism of B cells, naïve B cells are activated upon interaction of the surface immunoglobulin based B cell receptors with target antigens that are presented on MHC class II molecules [23]. There has been report of protective effects of antibodies in TB with IgA showing protection in mice [24]. However, IgG-based antibodies were found to mediate opsonization of MTb bacili for phagocytosis by macrophages [23]. The ability of IgA- and IgG-based antibodies to promote neutralization and confer protection enhances the importance of understanding antibody-based responses during TB infection [23]. Other human recombinant naïve libraries HAL7/8 were also applied to determine and characterized the specific antibodies against Mtb antigen 85B [25]. However, the HAL7/8 libraries are derived from healthy donors with a naïve repertoire. This makes the library useful for antibody generation for other targets but is not specifically suited for a particular disease.
In order to understand the extent of heightened antibody responses during TB infection, a systematic application to investigate the induced antibody responses in infected individuals would be required. Antibody phage display provides the unique ability to study the in vivo antibody representation using an in vitro approach [19]. Not only phage display, a combination between yeast and phage display was also used to select antibodies against Ag85, a TB biomarker [26]. Previously, an immune scFv library for TB was generated using TB-infected mice. The library was successfully used to generate scFvs against several TB proteins by panning [27]. Even so, the antibodies derived from the mouse immune library will not be useful for human therapeutics due to the immunogenicity issues [8]. Therefore, the development of human focused antibody library or immune antibody library derived from TB-infected individuals would help shed light on the extent of the antibody repertoire that is generated during TB infections in humans. In this study, a TB immune antibody library was generated using materials from six individuals infected with TB. The diversity required for immune library is lower, because of the presence of a skewed repertoire for disease-specific targets [5]. The application of materials derived from infected individuals will provide a better representation of the antibody repertoire as a response to the infection.
The quality of a library developed is mainly assessed based on the diversity of the library with the general rule that larger library sizes are more likely to provide higher affinity antibodies [28]. The library generated was of a modest diversity in the range 109, which can be considered satisfactory, as immune libraries normally require a lower diversity compared to naïve libraries to yield good quality antibodies [29]. Immune libraries of modest diversities around 105 have also been shown to produce high-affinity antibodies [30]. The V-gene repertoire of the library was focused on the IgG instead of IgA, although TB protection can be associated to the mucosal immunology. Even so, a comparison of antibody isotype responses toward TB has shown that IgG fractions are predominant for TB antigen fractions [31]. Further classification shows that IgG1 and IgG3 are most predominant subclasses in TB infections [32]. Therefore, the choice of isotype to be used for the generation of the immune library was IgG with the primer design covering the repertoire from all four IgG isotypes to provide a better coverage of IgG antibody responses against TB. This will hopefully increase the applicability of the library for antibody generation against other antigenic proteins of TB. The amplification of the repertoire was carried out individually to preserve the diverse repertoire from loss because of preferential amplification during PCR [33, 34].
The ensuing library was then used for biopanning, a standard approach used for the selection of antibodies against target antigens by affinity enrichment. After three rounds of selection against α-crystalline, approximately 63.6-fold enrichment of phage particles was obtained. The polyclonal phage ELISA result reflects the enrichment rate of the binders from each round with an increasing readout from rounds 1 to 3. Monoclonal antibody selection was selected using enriched phage from rounds 2 to 3. Although normal practices are to select for monoclonal antibodies mainly from the final round with the highest enrichment ratio, we opted to also include some clones from round 2. The reason was by selecting for clones from pre-high enrichment rounds, we wanted to increase the likelihood of obtaining clones of different identity and characteristics.
The panning and monoclonal selection was able to yield 26 binders to α-crystalline antigen, out of which only three clones presented complete sequences of the scFv without any unwanted mutations. The diversity of the antibody antigen-binding sites is attributed to the diversity of the complementarity-determining regions as well as by the synergistic influence of the VH and VL pairing. The three clones were unique in terms of the V-D-J segments used for the VH and V-D segments of the VL. There was a close similarity in the VH repertoire of clones 1H and 1B with VH2 and J3 being preferred. However, the diversifications of the D segment with D3–10*01 and D3–10*02 used for 1H and 1B, respectively. The main variation of the clones was in the VL pairing. Clone 1H was pairing with Kappa V3 and 1B with Kappa V4. Clone 10G was found to favor VH1 family with D4 and J6 gene segment in combination with Lambda 1 and J3 for the VL. The isolation of VH2 gene antibodies is normally rare but has been reported to be frequent in bacterial-associated infections [35]. Each VH and VL has three different CDR that have loop structure characteristic [36]. All three clones showed different CDR lengths that will have a direct impact on the topographical variation of the binding sites on the antibody [37]. Antibodies with grooves indicate peptide binders, while antibodies with “deep pockets” will bind to haptens [38]. The longer CDRs normally stabilized by cysteine-forming disulfide bridges [7]. The range for CDR3 length was between 13 to 22 aa for VH and 9 to 11 for VL.
All three antibody clones were successfully expressed and tested for its functionality in soluble form using ELISA. Although the purification of all three clones did not produce very high yields of soluble antibodies, all clones were able to generate a positive result in ELISA and Western blot using the soluble fractions. This indicates that the soluble antibody was still functional even in reduced conditions to bind to the antigen. The influences of VL pairing in the solubility and stability of antibodies have been reported to favor kappa light chains over lambda [39, 40]. This could be also a factor that influenced the isolation of two kappa clones during the panning experiment. Even so, the stability and solubility of the isolated lambda clone was good with comparable yield and binding characteristics shown by clone 10G.
The isolation of unique antibodies against a similar epitope from an immune library is not surprising. This is because the nature of the skewed diversity of the immune library would most likely result in antibody genes being diverted mainly toward an immunogenic site of the antigens [41]. This is because the immune repertoire would have already undergone affinity maturation processes to highlight preferential binding toward a particular epitope. Therefore, it is relevant to determine the cross-reactivity of antibodies isolated from a particular immune library to identify antibodies of different sequences that can target similar epitopes [7]. The competitive ELISA showed that clones 1B and 1H competes with each other for binding to the antigen. This could be due to the high degree of similarity in the V-D-J gene segment usage of the VH and VL of two clones. Although small pockets of variation in terms of certain gene segment usage were visible, this may have a profound effect on the overall affinity of the clones. Although the exact epitope region of the clones were not determined, however, we can conclude that the clones are either binding to the same epitope or are binding to sites in close proximity that could result in steric hindrance for competition to occur.
The application of phage display antibody libraries to study antibody responses in infectious diseases provides additional phenotypic information otherwise unattainable from solely the genotypic information. The success and variation in antibody clones enriched against the MTb α-crystalline target antigen highlights the impact TB infection has on antibody responses. Although the library was only applied against α-crystalline, the potential of using the library against other TB antigens may allow further understanding of the role antibodies play in TB immunology. A shortcoming to the study is the use of a small number of donors for the library construction. This may have a direct consequence on the representation of the V-gene repertoire analysis. Even so, this is a positive effort in correlating antibody-based responses as a result of TB infection. The library generated can be expanded in the future with more donor materials and applied against a host of other TB antigens to provide a deeper understanding on B cell responses during TB.
References
Mohajan, H. K. (2015). Tuberculosis is a fatal disease among some developing countries of the world. American Journal of Infectious Diseases and Microbiology, 3, 18–31.
Stewart, G. R., Robertson, B. D., & Young, D. B. (2003). Tuberculosis: A problem with persistence. Nature Reviews Microbiology, 1, 97–105.
Zumla, A., George, A., Sharma, V., Herbert, R. H. N., Baroness Masham of Ilton, Oxley, A., & Oliver, M. (2015). The WHO 2014 global tuberculosis report—further to go. The Lancet Global Health, 3, e10–e12.
Jacobs, A. J., Mongkolsapaya, J., Screaton, G. R., McShane, H., & Wilkinson, R. J. (2016). Antibodies and tuberculosis. Tuberculosis, 101, 102–113.
Lim, B. N., Tye, G. J., Choong, Y. S., Ong, E. B. B., Ismail, A., & Lim, T. S. (2014). Principles and application of antibody libraries for infectious diseases. Biotechnology Letters, 36, 2381–2392.
Kramer, R. A., Marissen, W. E., Goudsmit, J., Visser, T. J., Bakker, A. Q., de Jong, M., & Weldon, W. C. (2005). The human antibody repertoire specific for rabies virus glycoprotein as selected from immune libraries. European Journal of Immunology, 35, 2131–2145.
Rahumatullah, A., Ahmad, A., Noordin, R., & Lim, T. S. (2015). Delineation of BmSXP antibody V-gene usage from a lymphatic filariasis based immune scFv antibody library. Molecular Immunology, 67, 512–523.
Schroff, R. W., Foon, K. A., Beatty, S. M., Oldham, R. K., & Morgan, A. C. (1985). Human anti-murine immunoglobulin responses in patients receiving monoclonal antibody therapy. Cancer Research, 45, 879–885.
Beck, S. T., Leite, O. M., Arruda, R. S., & Ferreira, A. W. (2005). Humoral response to low molecular weight antigens of Mycobacterium tuberculosis by tuberculosis patients and contacts. Brazilian Journal of Medical and Biological Research, 38, 587–596.
Demkow, U., Zielonka, T., Nowak-Misiak, M., Filewska, M., Bialas, B., Strzalkowski, J., & Skopinska-Rozewska, E. (2002). Humoral immune response against 38-kDa and 16-kDa mycobacterial antigens in bone and joint tuberculosis. The International Journal of Tuberculosis and Lung Disease, 6, 1023–1028.
Yuan, Y., Crane, D. D., & Barry, C. E. (1996). Stationary phase-associated protein expression in Mycobacterium tuberculosis: function of the mycobacterial alpha-crystallin homolog. Journal of Bacteriology, 178, 4484–4492.
Teitelbaum, R., Glatman-Freedman, A., Chen, B., Robbins, J. B., Unanue, E., Casadevall, A., & Bloom, B. R. (1998). A mAb recognizing a surface antigen of Mycobacterium tuberculosis enhances host survival. Proceedings of the National Academy of Sciences, 95, 15688–15693.
Sixholo, J., Van Wyngaardt, W., Mashau, C., Frischmuth, J., Du Plessis, D. H., & Fehrsen, J. (2011). Improving the characteristics of a mycobacterial 16 kDa-specific chicken scFv. Biologicals, 39, 110–116.
Lim, T. S., Mollova, S., Rubelt, F., Sievert, V., Dübel, S., Lehrach, H., & Konthur, Z. (2010). V-gene amplification revisited—an optimised procedure for amplification of rearranged human antibody genes of different isotypes. New Biotechnology, 27, 108–117.
Lim, B. N., Chin, C. F., Choong, Y. S., Ismail, A., & Lim, T. S. (2016). Generation of a naïve human single chain variable fragment (scFv) library for the identification of monoclonal scFv against Salmonella Typhi Hemolysin E antigen. Toxicon, 117, 94–101.
Hairul Bahara, N. H., Chin, S. T., Choong, Y. S., & Lim, T. S. (2016). Construction of a semisynthetic human VH single-domain antibody library and selection of domain antibodies against α-crystalline of mycobacterium tuberculosis. Journal of Biomolecular Screening, 21, 35–43.
Brochet, X., Lefranc, M. P., & Giudicelli, V. (2008). IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized VJ and VDJ sequence analysis. Nucleic Acids Research, 36, W503–W508.
Lefranc, M. P., Giudicelli, V., Kaas, Q., Duprat, E., Jabado-Michaloud, J., Scaviner, D., & Lefranc, G. (2005). IMGT, the international ImMunoGeneTics information system®. Nucleic Acids Research, 33, D593–D597.
Retter, I., Althaus, H. H., Münch, R., & Müller, W. (2005). VBASE2, an integrative V gene database. Nucleic Acids Research, 33(Database issue), D671–D674.
Finlay, W. J., & Almagro, J. C. (2012). Natural and man-made V-gene repertoires for antibody discovery. Frontiers in Immunology, 3, 342.
Larimore, K., McCormick, M. W., Robins, H. S., & Greenberg, P. D. (2012). Shaping of human germline IgH repertoires revealed by deep sequencing. The Journal of Immunology, 189, 3221–3230.
Wu, L., Oficjalska, K., Lambert, M., Fennell, B. J., Darmanin-Sheehan, A., Shúilleabháin, D. N., & Paulsen, J. (2012). Fundamental characteristics of the immunoglobulin VH repertoire of chickens in comparison with those of humans, mice, and camelids. The Journal of Immunology, 188, 322–333.
Rao, M., Valentini, D., Poiret, T., Dodoo, E., Parida, S., Zumla, A., Brighenti, S., & Maeurer, M. (2015). B in TB: B cells as mediators of clinically relevant immune responses in tuberculosis. Clinical Infectious Diseases, 61(Suppl 3), S225–S234. https://doi.org/10.1093/cid/civ614.
Williams, A., Reljic, R., Naylor, I., Clark, S. O., Falero-Diaz, G., Singh, M., & Ivanyi, J. (2004). Passive protection with immunoglobulin A antibodies against tuberculous early infection of the lungs. Immunology, 111, 328–333.
Fuchs, M., Kämpfer, S., Helmsing, S., Spallek, R., Oehlmann, W., Prilop, W., & Hust, M. (2014). Novel human recombinant antibodies against mycobacterium tuberculosis antigen 85B. BMC Biotechnology, 14, 68.
Ferrara, F., Naranjo, L. A., Kumar, S., Gaiotto, T., Mukundan, H., Swanson, B., & Bradbury, A. R. (2012). Using phage and yeast display to select hundreds of monoclonal antibodies: application to antigen 85, a tuberculosis biomarker. PloS One, 7, e49535.
Cummings, P. J., Hooper, N. E., & Rowland, S. S. (1998). Generation of a recombinant bacteriophage antibody library to Mycobacterium tuberculosis. Hybridoma, 17, 151–156.
Schaffitzel, C., Hanes, J., Jermutus, L., & Plückthun, A. (1999). Ribosome display: an in vitro method for selection and evolution of antibodies from libraries. Journal of Immunological Methods, 231, 119–135.
Moon, S. A., Ki, M. K., Lee, S., Hong, M. L., Kim, M., Kim, S., & Shim, H. (2011). Antibodies against non-immunizing antigens derived from a large immune scFv library. Molecules and Cells, 31, 509–513.
Chassagne, S., Laffly, E., Drouet, E., Hérodin, F., Lefranc, M. P., & Thullier, P. (2004). A high-affinity macaque antibody Fab with human-like framework regions obtained from a small phage display immune library. Molecular Immunology, 41, 539–546.
Perley, C. C., Frahm, M., Click, E. M., Dobos, K. M., Ferrari, G., Stout, J. E., & Frothingham, R. (2014). The human antibody response to the surface of Mycobacterium tuberculosis. PloS One, 9, e98938.
Sousa, A. O., Henry, S., Maroja, F. M., Lee, F. K., Brum, L., Singh, M., & Aucouturier, P. (1998). IgG subclass distribution of antibody responses to protein and polysaccharide mycobacterial antigens in leprosy and tuberculosis patients. Clinical and Experimental Immunology, 111, 48–55.
Suzuki, M. T., & Giovannoni, S. J. (1996). Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Applied and Environmental Microbiology, 62, 625–630.
Kanagawa, T. (2003). Bias and artifacts in multitemplate polymerase chain reactions (PCR). Journal of Bioscience and Bioengineering, 96, 317–323.
Andris, J. S., Brodeur, B. R., & Capra, J. D. (1993). Molecular characterization of human antibodies to bacterial antigens: utilization of the less frequently expressed VH2 and VH6 heavy chain variable region gene families. Molecular Immunology, 30, 1601–1616.
Chothia, C., & Lesk, A. M. (1987). Canonical structures for the hypervariable regions of immunoglobulins. Journal of Molecular Biology, 196, 901–917.
Kunik, V., & Ofran, Y. (2013). The indistinguishability of epitopes from protein surface is explained by the distinct binding preferences of each of the six antigen-binding loops. Protein Engineering Design and Selection, 26, 599–609.
Marchuk, D., Drumm, M., Saulino, A., & Collins, F. S. (1991). Construction of T-vectors, a rapid and general system for direct cloning of unmodified PCR products. Nucleic Acids Research, 19, 1154.
Ewert, S., Huber, T., Honegger, A., & Plückthun, A. (2003). Biophysical properties of human antibody variable domains. Journal of Molecular Biology, 325, 531–553.
Kim, D. Y., To, R., Kandalaft, H., Ding, W., van Faassen, H., Luo, Y., & Kelly, J. F. (2014). Antibody light chain variable domains and their biophysically improved versions for human immunotherapy. In MAbs, 6, 219–235.
Georgiou, G., Ippolito, G. C., Beausang, J., Busse, C. E., Wardemann, H., & Quake, S. R. (2014). The promise and challenge of high-throughput sequencing of the antibody repertoire. Nature Biotechnology, 32, 158–168.
Acknowledgements
The authors would like to acknowledge the support from the Malaysian Ministry of Higher Education through the Fundamental Research Grant (FRGS) Scheme (grant no: 203/CIPPM/6711381) and Malaysian Ministry of Higher Education Higher Institution Centre of Excellence (HICoE) grant (grant no: 311/CIPPM/44001005). NMN acknowledges the support from the Malaysian Ministry of Higher Education through the Long-term Research Grant Scheme (LRGS) Scheme (grant no: 203/PPSK/67212002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Hamidon, N.H., Suraiya, S., Sarmiento, M.E. et al. Immune TB Antibody Phage Display Library as a Tool To Study B Cell Immunity in TB Infections. Appl Biochem Biotechnol 184, 852–868 (2018). https://doi.org/10.1007/s12010-017-2582-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-017-2582-5