
Abstract
Granulocyte colony-stimulating factor (GCSF) has therapeutic applications due to its proven efficacy in different forms of neutropenia and chemotherapy-induced leucopenia. The original 564-bp nucleotide sequence from NCBI was codon optimized and assembled by overlapping PCR method comprising of 16 oligos of 50-nt length with 15 base overhang. The synthetic gene (CO-GCSF) was cloned under glucose utilizing glyceraldehyde 3-phosphate dehydrogenase (GAP) and methanol-utilizing alcohol oxidase (AOX1) promoters and expressed in Pichia pastoris SMD1168 strain. Constitutive expression under GAP resulted in cellular toxicity while AOX1 promoter controlled expression was stable. Variation in the levels of expression was observed among the transformant colonies with transformant #2 secreting up to ∼4 mg/L of GCSF. The molecular mass of the expressed GCSF in P. pastoris was ∼19.0 kDa. Quatitation of the expressed protein was carried out by a highly reproducible gel densitometric method. Effect of several operational and nutritional conditions was studied on GCSF production and the results suggest a general approach for increasing the yield of GCSF several folds (2- to 5-fold) over the standard conditions employed currently. Cultivation of the single-copy integrant in the chemically defined medium in a 5-L fermenter resulted in a volumetric productivity of ∼0.7 mg/L/h at the end of the induction phase, which was about 4-fold higher than attained in the shake flask.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Human granulocyte colony-stimulating factor (GCSF) has therapeutic applications and is a hematopoietic growth factor. Its efficacy has been proven in different forms of neutropenia and chemotherapy-induced leucopenia, one of the major side effects of chemotherapy and radiotherapy. Produced by monocytes and macrophages, it stimulates proliferation, activation, and differentiation of precursor cells through JAK-STAT signal transduction pathway [1]. It has been shown that GCSF stimulates mobilization of progenitor cells for autologous or allogenic transplantations [2]. It also finds applications after bone marrow transplantation and in AIDS therapy [3]. The natural human mature glycoprotein exists in two forms, a 174- and 177-amino acid long protein, of which the 174-amino acid form is the pharmaceutically relevant type [4].
Two forms of the recombinant human GCSF are currently in commercial use and these are the recombinant non-glycosylated GCSF from Escherichia coli and the other is derived from mammalian expression system of Chinese hamster ovarian (CHO) cell line, which is O-glycosylated at Thr-133 position [5]. The former is sold as filgrastim (Neupogen®) and its structure differs slightly from the structure of the natural glycoprotein. The pegylated or PEG (polyethylene glycol)-filgrastim (Neulasta®) is again the E. coli-produced enzyme which has a much longer half-life, reducing the necessity of daily injections. The GCSF derived from the CHO cells is indistinguishable from the natural human GCSF. Although glycosylation is not necessary for biological activity of this protein, it confers stability by suppressing the polymerization and conformational change [6]. It is also reported [7] to lend stability to GCSF towards proteolysis. Given the fact that the cost of production determines the final cost of the product, Pichia pastoris offers a good alternative that combines the ease of production and post-translational modifications akin to higher eukaryotic systems. For less complex proteins, it has been reported that the efficiency of the P. pastoris system is better due to shorter processing time and higher biomass density leading to higher space-time yield of the proteins [8]. Already, one sixth of the approved therapeutics are produced in yeasts which include Saccharomyces cerevisiae and Hansenula polymorpha.
P. pastoris harbors several strong promoters that can be exploited to drive heterologous expression of recombinant genes, both in a constitutive or inducible fashion, and a number of therapeutic proteins have been successfully expressed in this system. The cells can be cultivated to high densities in chemically defined medium and extracellular protein levels to the extent of 6 g/L have been reported [9]. Different feeding strategies [10], as well as a combination of methanol and other carbon sources [11] during the induction phase, have been reported to increase productivities by several folds. Absence of contaminating extracellular native proteins also helps in purification of the heterologous proteins from culture filtrate. There are several reports on expression of GCSF in the P. pastoris system with extracellular levels ranging from 3.1 mg/L [12] for the non-optimized gene to 18 mg/L for codon-optimized copy [13]. Aggregation of GCSF has been widely reported in the Pichia system [13–15] and the renatured form could be recovered after denaturation with urea and guanidine HCl leading to higher yields. Other strategy included addition of surfactants during induction [15] which resulted in a yield of 200–250 mg/L in shake flask studies.
In the present study, a codon-optimized synthetic copy of GCSF was used to improve the yield. It is a well-established fact that codon usage pattern regulates both expression and folding patterns in heterologous systems [16]. The synthetic gene was expressed as a fusion protein with the α-factor signal peptide and expressed under the control of glyceraldehyde 3-phosphate dehydrogenase (GAP) promoter for constitutive expression and alcohol oxidase 1 (AOX1) promoter for inducible expression. A number of factors commonly reported [17] to affect heterologous expression in this yeast were investigated and some factors were identified. Expression was also studied in a 5-L fermenter to arrive at conditions leading to high productivity and stability of the product.
Materials and Methods
Plasmids and Host Strains
A codon-optimized GCSF synthetic gene (developed as shown below) was cloned into multiple cloning site of commercially available expression vectors pGAPZαA and pPICZαB (Invitrogen). E. coli DH5α was used as the host strain for propagating the constructed plasmids. The transformants were cultivated in Luria-Bertani agar plates containing 100 μg/mL zeocin. P. pastoris GS115 (Invitrogen) and SMD1168 (his4pep4, Invitrogen) were used as host strains which were maintained on yeast extract peptone dextrose medium (YPD).
GCSF Open Reading Frame Design and Construction
The protein sequence of human GCSF was retrieved from gene database (GenBank accession no. NP_757373.1). The open reading frame was generated based on protein sequence of isoform-b of human GCSF molecule of 174 aa residues. The synthetic gene excluded the first 30 aa of the native signal peptide. The protein sequence was submitted to a software program, Premier Biosoft, by which nucleotide sequence of synthetic GCSF gene was generated. The synthetic GCSF gene was optimized to codon preference for P. pastoris. To construct this synthetic GCSF gene in vitro, 16 primers were used, each having 50 nucleotides with an overlapping region of 15 base pairs between adjacent primers. XbaI and XhoI sites were included in the 5′ and 3′ terminal primers, respectively, for cloning into the expression vector.
The synthetic GCSF construct was assembled in a stepwise fashion using multiple PCR reactions (for process outlay, see Fig. 1). The gene synthesis was in three stages, stage 1 and stage 2 were divided in to two sets. In stage 1, four separate PCR reactions were set up with respective primer pairs for each set (set 1 primer pairs: FP1-RP1, FP2-RP2, FP3-RP3, and FP4-RP4 and set 2 primer pairs: FP5-RP5, FP6-RP6, FP7-RP7, and FP8-RP8) (Supplementary Fig. 1). Representative sequences for four primers are shown in Table 1. The products of the first four PCR reactions of set 1 were annealed to each other one by one to synthesize a mega construct 1 (MC1). Similarly, mega construct 2 (MC2) was synthesized from set 2 PCR reactions. In stage 2, mega constructs were amplified using PCR reaction (set 1 primer pair: FP1-RP4 and set 2 primer pair: FP5-RP8). In stage 3, mega constructs (MC1 and MC2) were mixed, annealed, and amplified by PCR using FP1-RP8 as primer pair to get final sequence of the synthetic GCSF gene. The product obtained was purified by gel extraction and further amplified by PCR to get a product rich in the synthetic GCSF gene. The sequence of the codon-optimized synthetic GCSF construct (CO-GCSF) was confirmed by sequencing. The nucleotide sequence of GCSF was re-encoded into codons to confirm the correct amino acids. Any changes noticed were corrected by site-directed mutagenesis.

Process outlay for assembling synthetic codon-optimized GCSF gene through oligo assembly
Cloning and Transformation of Host Strains
The CO-GCSF synthetic gene was cloned in to XhoI–XbaI sites of the vectors pGAPZαA and pPICZαB in-frame with the α-factor signal sequence to generate pGAPZαA-CO-GCSF and pPICZαB-CO-GCSF. The details of the construct in pPICZαB are given in Fig. 2. The recombinant vectors were transformed in to competent E. coli DH5α cells for propagation of the plasmids and the constructs were confirmed by sequencing. As a control, native human cDNA (devoid of leader peptide) was cloned in pPICZαB in-frame with the α-factor signal sequence. All the constructed expression vectors were linearized with SmaI and transformed in to competent P. pastoris strains GS115 and SMD 1168 by electroporation (2000 V/cm, 25 μF, 400 Ω; Gene Pulser Electroporation System, Bio-Rad Laboratories, Hercules, CA, USA). After pulsing, electro-competent cells were placed in ice-cold 1 M sorbitol (1 mL) and regenerated at 30 °C for 2 h. Transformants were plated (100 μL) on yeast extract peptone dextrose sorbitol medium containing 100 μg/mL zeocin and grown at 30 °C for 3–5 days. The zeocin-resistant transformants were screened for the presence of the insert by colony PCR. Methanol utilizing capacity was determined by plating out on methanol containing medium (Invitrogen). The transformants were grown in shake flasks to observe the expression level of GCSF.

An outlay of the vector pPICZαB showing the site of ligation of the synthetic codon-optimized GCSF gene
GCSF Production in Shaking Flasks
The positive transformants were tested for their ability to produce GCSF in the culture medium. The transformants using the GAP promoter (GAP/CO-GCSF) were cultivated in 10-mL YPD medium for 72 h with shaking at 28 °C. One milliliter of sample was collected every 24 h to monitor growth, extracellular protein, and GCSF production (after concentration by 10-fold, see below). For transformants using the AOX1 promoter (AOX1/CO-GCSF), cultivation was carried out initially in 10 mL of YPD medium (1 % yeast extract, 2 % peptone, and 2 % dextrose) contained in 100 mL of flask and grown overnight at 30 °C with shaking at 230 rpm. The overnight cultures were transferred to a 100-mL buffered glycerol-complex medium (1 % yeast extract, 2 % peptone, 1.34 % YNB, 1 % glycerol, 100 mM phosphate buffer pH 6.0, 4 × 10−5 % biotin, 0.004 % histidine) in a 500-mL flask. After growing at 28 °C in a shaking incubator (230 rpm) overnight, the cell pellets were harvested by centrifugation (3000g, 5 min, 25 °C) and re-suspended in a 100-mL buffered methanol-complex medium (BMMY) containing 1 % yeast extract, 2 % peptone, 1.34 % YNB, 1 % methanol, 100 mM phosphate buffer pH 6.0, 4 × 10 − 5 % biotin, 0.004 % histidine) in 500-mL baffled flask (production flasks). One milliliter of 100 % methanol was added every day to induce expression. To analyze protein expression following methanol induction, culture supernatants were collected every 24 h for 4 days.
For the selected clone# 2 (AOX1/CO-GCSF), production of GCSF was investigated at different temperatures (24 and 28 °C) and initial pH (4, 5, 6, and 7) during the induction phase. Effect of varying methanol concentration from 0.5–2 %, during induction phase, was also studied. Supplementation with, sorbitol (as an additional carbon source) and a mixture of amino acids (Ala, Gln, Gly, Leu, and Ser, each at 20 mg/100 mL) was investigated on GCSF production by their addition in the beginning of the methanol induction phase. Samples were removed 4 days after the start of the induction phase with methanol added at 1 % every 24 h to maintain induction of the AOX1 promoter. Cell O.D., extracellular protein, and GCSF levels were measured in the culture filtrate.
Bench Scale Fermentation and Production of GCSF in AOX1/CO-GCSF Transformant #2
The transformant #2 was cultivated in a 5-L bioreactor (Applikon, the Netherlands) in fed-batch mode using chemically defined medium. The composition of the medium was as follows (g/L): (NH4)2SO4, 30; KH2PO4, 12.6; Na2HPO4·2H2O, 8.22; glycerol, 24; MgSO4·7H2O, 7. Trace element solution was added as per standard protocols at 4.5 mL/L of the medium. Batch phase was started using 1.3 L of the autoclaved medium containing 2.4 % (v/v) of glycerol. The clone was passaged through YPD medium in to 100 mL of defined medium in 0.5-L baffled shake flask. After overnight growth in the medium to an OD600 of ∼20, a part of it was inoculated in to the fermenter giving an initial OD600 of 0.2–0.3. Aeration was set at 0.15 L/min (0.1 vvm). Agitation was initially set to 600 and later increased to 1200 rpm in steps to maintain dissolved oxygen levels at >30 % of saturation. Pure oxygen was purged automatically as and when required. After exhaustion of glycerol, fed-batch was started by pumping feed 1 (containing 50 % glycerol + trace element solution) to maintain specific growth rate of 0.11 h−1. After feed 1 (FB-1), a transition phase of 12 h was given for adaptation to methanol in a mixture of methanol and sorbitol along with trace element solution by adding feed 2 (FB-2). The pH was maintained at 5.5 ± 0.05 by addition of 15 % (v/v) ammonia solution which also served as a nitrogen source. Methanol induction phase was maintained by supplying feed 2 at a rate to maintain specific growth rate at 0.02 h−1. Antifoam A concentrate (Sigma Chemical Co., St. Louis, MO) (3 drops) was added prior to inoculation, and as needed thereafter, to prevent excessive foaming. Temperature was maintained at 28 °C during the entire period of fermentation. Samples were taken at regular intervals to determine cell growth, extracellular protein, residual methanol, sorbitol, and GCSF level.
Analytical Methods
Cell growth was monitored by measurement of OD at 600 nm and packed cell volume. Total proteins in the culture supernatant were precipitated by TCA-acetone precipitation method. The protein pellet was solubilized in solubilization buffer (62.5 mM Tris-Cl, pH 6.8; 1 % SDS, 6.25 % β-mercaptoethanol). Proteins were quantified by Bradford method using BSA as a standard. Proteins were separated in 12 % SDS-PAGE gels according to Laemmli and the gels stained with Coomassie Brilliant Blue R-250. The identity of the produced GCSF band in the gel was confirmed by MALDI-TOF analysis of the extracted protein band.
Quantitation of GCSF by Densitometric Method and ELISA
The GCSF content was determined by densitometric analysis by running the commercial filgrastim (0.6–3.0 μg/lane) on 12 % polyacrylamide gels along with the concentrated culture supernatants. The gels were stained with Coomassie Brilliant Blue R-250 and read using Quantity One software in Gel documentation unit (Bio Rad). The concentration of GCSF was calculated from the standard graph. GCSF concentration in concentrated culture supernatant was also assayed using self sandwich ELISA method. Briefly, a rabbit polyclonal antibody IgG (SantaCruz Biotechnology) representing full-length GCSF (207 aa) was coated (100 μL/well) on the 96 well micro-titer plate (Geneticin) at 4 °C overnight. The plate was washed three times with phosphate-buffered saline (PBS, 137 mM NaCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, pH 7.4) and then blocked with 200 μL/well of 1 % (w/v) BSA in PBS at 37 °C for 4 h with gentle shaking. After washing, serially diluted GCSF standards (100 μL) and culture supernatant (50 μL) appropriately diluted in PBS were added to each well, and the plates were incubated at 37 °C for 3 h. After washing, 100 μL of rabbit polyclonal antibody IgG, diluted in PBS, was added as the primary antibody and incubated at 37 °C for 30 min. After washing, 100 μL of alkaline phosphatase (AP)-conjugated goat anti-rabbit immunoglobulin G, diluted in PBS, was added as the secondary antibody and incubated at 37 °C for 1 h. After washing, 50 μL of the substrate solution containing p-nitro-phenyl-pyrophosphate was used to visualize the signal. The reaction was stopped after 20 min with 2 N NaOH. The plates were analyzed at 420 nm using a 96-well microplate reader (SpectraMax M2®, Molecular Devices LLC, CA, USA).
Statistical Analysis
During the screening part for high-yielding transformants, experiments were conducted in triplicates two times. Studies on the transformant #2 were carried out in three independent experiments run in duplicates. The variation within the experiments was between 7–10 %.
Results and Discussion
Construction of Synthetic Gene and Isolation of Transformants Using pGAPZαA-CO-GCSF and pPICZαB-CO-GCSF
The full length of human GCSF cDNA is 564 bp encoding a polypeptide of 174 aa with a signal sequence. In the present study, the 522-bp fragment coding for mature GCSF was used for codon optimization based on the P. pastoris preference for codons. The details of the codon used are given in Table 2. The sequence alignment results showed that the codon-optimized (CO-) GCSF shared ∼80 % identity with the human cDNA of GCSF. The gene was assembled from short DNA fragments assembled piece by piece. A total of 16 small DNA fragments were put together, raised through oligos of 50-nt length and a 15-bp overlapping region. The MC1 and 2 were finally assembled to build the full length copy of the gene with flanking XhoI–XbaI sites for cloning in to two vectors. The sequence of both the constructs was verified. Two nucleotide mismatch amplifications, occurring during PCR reactions, were corrected by in vitro mutagenesis. The vector allows for the integration event at the AOX1 site in the genome and growth on zeocin plates. The integration of the gene was confirmed by colony PCR with GCSF-specific primers. Almost all the transformants obtained with the two vectors gave a PCR product verifying the integration event.
Expression of GCSF in Shake Flask
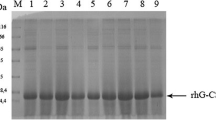
The GAP promoter driven expression of CO-GCSF was found to be toxic and the cell OD600 never crossed a value of 8 OD units. GAP driven expression has been reported to be effective for certain proteins [18], and level of heterologous protein to the extent of 250 mg/L was obtained. In many other studies, however, GAP-controlled constitutive expression has been indicated to cause cell toxicity due to constitutive expression of the heterologous protein [19]. Thus, inducible expression of CO-GCSF was next attempted in P. pastoris GS115 using the AOX1 promoter. In this host, a cell OD600 of about 30 was achieved after 96 h of cell growth. Protein analysis, after TCA-acetone precipitation of culture supernatant proteins, on 12 % SDS-PAGE indicated expression to the extent of a few milligrams per liter but GCSF was found to be unstable in the culture filtrate. Thus, AOX1-driven expression was next investigated in SMD1168 (protease negative) strain. A number of PCR-positive transformants (#s 1–12) were picked up and cultivated in BMMY medium for screening of high GCSF-producing clone. It has been reported that even during a single transformation event, a considerable variation exists in the level of production of the heterologous proteins in the transformants and thus a number of individual colonies need to be screened [20]. The results obtained with the 12 positive clones are shown in Fig. 3a. As seen, the final cell O.D. varied between 18 to 48 and no clear correlation could be observed between the cell growth and GCSF production. The quatitation by ELISA indicated low levels of GCSF production in the culture supernatant with maximum value of 40 μg/L in transformant #2. Another approach, using the gel densitometric tracing, was developed for quantitation of GCSF since it is reported (ELISA technical guide and protocols, Thermo Scientific, http://www.piercenet.com/method/overview-elisa) that using the same antibody as both capture and detection antibodies in self-sandwich ELISA can be less efficient in terms of its dynamic range and binding of the entire antigen of small molecular weight. This is due to steric hindrances posed to detection antibody and cross reactivity of the capture and secondary antibody. The gel densitometric method was found to be suitable with a consistently obtained linear relationship in the concentration range of 0.6- to 3.0-μg GCSF (Supplementary Fig. 2). The SDS-PAGE analysis of the concentrated extracellular proteins (precipitated by TCA-acetone method) was carried out for the transformants and the data indicated maximum production in transformant #2. Such differences in the level of production among transformants have been attributed to altered methanol utilization as a result of different transformation events. A time course profile of GCSF production in clone #2 is shown in Fig. 3b. A major protein band at ∼19 kDa was observed which was absent in the control (vector alone) sample. The higher molecular mass was attributed to GCSF being glycosylated in the Pichia secretory pathway. While no N-linked glycosylation is possible in GCSF, there are four putative O-glycosylation sites (NetOGlyc 4.0 server), out of which the glycosylation at Thr 122 is the most relevant one [15]. GCSF was quantitated using the standards run on the gel under identical conditions and an overall level of ∼4 mg/L was produced in the original culture filtrate at the end of 24 h post-induction with methanol, much earlier than that reported for the non-codon-optimized levels in other studies [12, 15]. This level was also 6-fold higher than that observed with the native cDNA expressed GCSF in this host. It may be attributed to increased metabolic burdens on intracellular processes due to incorrect/slow folding of the protein arising out of the translation of the non-native cDNA. A number of studies confirm that replacement of codons with the host preferred codons affects both stability and translation rates thereby affecting stability and secretion [16]. A productivity of 0.17 mg/L/h was obtained in the shake flask and is the highest reported so far. The protein band produced was confirmed as GCSF by MALDI-TOF analysis of the tryptic fragments (see Supplementary Fig. 3).

a Screening of SMD1168 transformants for GCSF production: bar chart comparing final cell O.D. and GCSF levels for various transformants (#1–12 and control containing only vector). b SDS-PAGE analysis of 25-x concentrated total proteins in culture medium of P. pastoris transformant #2 showing GCSF production. Lane 1: standard GCSF. Lane 2: molecular marker lane. Lanes 3 and 4: control (empty vector). Lanes 5–8: samples at 24, 48, 72, and 96 h post-induction with methanol. Arrow indicates GCSF band
Effect of Tween Addition, Process Parameters, and Supplementary Nutrients on GCSF Production
It has been reported [15] that addition of Tweens can enhance extracellular release of GCSF in P. pastoris due to an increase in membrane permeability on account of varying hydroxyl contents. However, in the present study, no effect of Tweens (20, 40, 60, and 80) was observed on secretion efficiency of the SMD 1168 host. Effect of lowering the temperature during the production phase was also investigated but no significant increase in extracellular GCSF production was attained. Significant changes in the yield of heterologous proteins have been reported as a function of temperature in various studies [21, 22] which has been attributed to suppression of protein misfolding at lower temperature thereby facilitating secretion. During expression of 20 mammalian G protein-coupled receptors in P. pastoris [23], enhancement in expression was observed for some proteins but had no effect on several other receptors indicating the effect to be protein dependent. Our own studies with a large 90-kDa β-glucosidase I production in P. pastoris indicated temperature to have a significant effect and nearly 2-fold higher activity was obtained at a temperature of 24 °C when compared to 28 °C [24].
Stability of the secreted proteins in the extracellular culture filtrate of P. pastoris is dependent on the protease action and its ability to retain its folding in the environment. pH exerts a considerable influence on both of these factors. Each protein due to properties inherent to its sequence and structure has different stability. P. pastoris has a wide range of pH tolerance from 3–9. Though most of recombinant proteins are produced in range of 5–6, there are specific cases where much higher or lower pH values were required. For instance, production of influenza A virus hemagglutinin protein was found to be highest at pH 8.0 owing to its higher stability [25] and a pH of 7.5 favored high production of β-glucosidase I [24]. Lower pH optimum (2–3.5) has been reported for production of insulin-like growth factor due to proteolytic degradation at higher pH [26] and pH of 3.0 was found to be optimum for recombinant gelatin [27]. In the present study, initial pH of 4 and 5 increased GCSF production by 5-fold and 3-fold, respectively, while no effect was observed on cell density. It was attributed to either increased stability of GCSF at low temperature, as reported previously [28] or that citrate ions used for preparation of the low pH buffer served as additional carbon source for biomass accumulation and thereby product formation.
Based on a previous observation [29] that addition of specific amino acids, particularly those that occur with high frequency in the protein to be expressed, in the culture medium-enhanced heterologous protein production, this strategy was adopted in the present study. The amino acid distribution in GCSF was calculated from the coding sequence and five amino acids occurring at high frequency were identified. These were Ala (11.8 %), Gln (9.8 %), Gly (7.4 %), Leu (19.1 %), and Ser (7.4 %). Supplementation with a mixture of these amino acids in the BMMY medium resulted in 2.2-fold increase in extracellular GCSF levels over the unsupplemented cultures. A similar 2-fold increase was observed during production of a model protein in P. pastoris system [30].
For genes expressed under the control of the AOX1 promoter, optimization of methanol levels are expected to play an important role. The higher the methanol concentration, the higher will be the recombinant protein production, but reports point out that while a low methanol concentration may not be adequate enough to trigger the transcription, a higher methanol concentration or accumulation could lead to accumulation of toxic formaldehyde causing cell lysis and release of proteases [17]. This study pointed out the optimum level of methanol to be 1 %. In view of the observations that presence of additional carbon source, during the methanol induction phase, can lead to increase in cell biomass without accumulation of toxic by-products of methanol metabolism and low heat generation [31], sorbitol was selected. About a 2-fold increase in GCSF was noted. A summary of the major effects observed on account of various nutritional and process parameters is shown in Fig. 4. Maximum level of GCSF (18 mg/L) was attained when the initial pH of the production phase was set at 4.0 giving a productivity of 0.75 mg/L/h, highest reported so far (Table 3) without the addition of Tweens to which different hosts respond differently.

Summary of effects of process parameter and various supplementations on final biomass (cell O.D.) and GCSF levels (fold increase is calculated with respect to reference conditions, i.e., BMMY medium with 1 % v/v methanol feeding, pH 6.0, and cultivation temperature of 28 °C)
Production of GCSF in a Bioreactor
Fermenter run performed with complex medium indicated less extracellular protein in the culture broth with excessive foaming resulting in denaturation and low productivity of GCSF. Accordingly, the fermenter was operated using chemically defined medium. This strategy has been adopted by several workers for large-scale fermentation using the P. pastoris system (for review on the subject, see [32]). As observed in Fig. 5a, the batch phase lasted for 36 h followed by derepression and a transition phase wherein adaptation to methanol occurred by a mixed feed of methanol and sorbitol. This mixed feeding was necessary during the induction phase to increase the cell O.D. which could not increase beyond 80 when supported on methanol alone. At the end of feed 1, cell O.D. increased from ∼80 to 220 indicating a buildup of biomass. A gradual increase in extracellular protein to 300 mg/L was observed at the end of the induction phase (75 h after addition of mixed feed). Gel data (Fig. 5b) indicated steady level of GCSF in the culture broth and a productivity of 0.71 mg/L/h and a yield of 0.66 mg/g cell dry wt. This was about 0.2-fold higher than the value attained in the shake flask. This level of productivity was maintained until cell harvest. The biological efficacy of the P. pastoris produced GCSF has been reported previously (15). Analysis of the cell morphology/survival carried out at different stages indicated budding cells at all stages with >90 % survival rate. SDS-PAGE analysis confirmed stability of the product as no hydrolysis was noticed of the synthesized GCSF. A number of studies have been conducted on fed-batch fermentation of the P. pastoris system and it has been shown that specific secretion rate correlates well with specific growth rate, μ [33]. Thus, optimization studies in the Pichia system are based on initial high feed rate for biomass accumulation followed by a phase of decreasing μ, thus allowing for product accumulation. Such a strategy leads to a shorter processing time and gives a high space-time yield. In the present study, this strategy worked well and allowed for higher product outcome per bioreactor volume and time. This is relatively significant as the capital cost in the biopharmaceutical industry is high [34] and maximizing productivity is an important process strategy. It is also important to note that the product produced was stable and did not aggregate and it is expected to simplify the downstream processing operations.

a GCSF production in a 5 L-fermenter in chemically defined medium at reference conditions (pH 6.0, 28 °C) supplemented with methanol and sorbitol feed. Cell O.D., pH, DO, extracellular protein, and GCSF are shown at different time points. b SDS-PAGE analysis of total proteins produced by transformant #2 in a 5 L-fermenter (lane 1: medium-range protein markers; lane 2: standard GCSF, 0.9 μg; lanes 3–7: 20 μL culture supernatant from sampling time of 72, 84, 96, 108, and 122 h of total fermentation time. Arrow indicates the GCSF band
Conclusions
In summary, a codon-optimized copy of GCSF, expressed under the control of the AOX1 promoter in the P. pastoris SMD 1168 host exhibited a considerable heterogeneity in production levels among different transformants. Maximum productivity of 0.16 mg/L/h was exhibited by transformant #2. The level of protein produced in this transformant was 6-fold higher than that produced by the native cDNA-directed GCSF. The production was affected by initial pH of the production phase with an increase in absolute levels by 5- and 3-fold at pH of 4 and 5, respectively. Supplementation with a mixture of amino acids increased the level of GCSF by 2.2-fold. A maximum yield of 18 mg/L was achieved at the end of 24 h when an initial pH of 4.0 was used during the induction phase. Although a somewhat similar level (15 mg/L) was reported earlier [35] in P. pastoris SMD 1168 strain, the productivity was ∼5-fold lower than that obtained in the present study. Mixed carbon feed during the induction phase lead to a higher biomass accumulation and an increase in productivity which was implemented in fed-batch fermentation. A productivity of 0.71 mg/L/h was achieved in the bioreactor in the induction phase. No aggregation or proteolytic degradation was observed of the synthesized product at shake flask or reactor level. Given various genetic and process strategies that can be implemented in P. pastoris, high yield and productivity can be obtained for this protein.
References
Biethahna, S., Alvesa, F., Wildea, S., Hiddemanna, W., & Spiekermann, K. (1999). Expression of granulocyte colony-stimulating factor and granulocyte-macrophage colony stimulating factor associated signal transduction proteins of the JAK/STAT pathway in normal granulopoiesis and in blast cells of acute myelogenous leukemia. Experimental Hematology, 27, 885–894.
Viret, F., Goncalves, A., Tarpin, C., Chabannon, C., et al. (2006). GCSF in oncology. Bulletins Cancer, 93, 463–471.
Bell, E. (2009). Transplantation: GCSF therapy after BMT: getting the timing right. Nature Reviews in Immunology, 9, 308–309.
Nagata, S., Tsuchiya, M., Asano, S., Yamamoto, O., Hirata, Y., Kubota, N., Oheda, M., Nomura, H., & Yamazaki, T. (1986). The chromosomal gene structure and two mRNAs for human granulocyte colony stimulating factor. EMBO Journal, 5, 575–581.
Kubota, N., Orita, T., Hattori, K., Oh-eda, M., et al. (1990). Structural characterization of natural and recombinant human granulocyte colony-stimulating factors. Journal of Biochemistry, 107, 486–492.
Oh-eda, M., Hasegawa, M., Hattori, K., Kuboniwa, H., et al. (1990). O-linked sugar chain of human granulocyte colony-stimulating factor protects it against polymerization and denaturation allowing it to retain its biological activity. Journal of Biological Chemistry, 265, 11432–11435.
Carter, C. R. D., Whitmore, K. M., & Thorpe, R. (2004). The significance of carbohydrates on G-CSF: differential sensitivity of GCSFs’ to human neutrophil elastase degradation. Journal of Leukocyte Biology, 75, 515–522.
Maccani, A., Landes, N., Stadlmayr, G., Maresch, D., Leitner, C., Maurer, M., Gasser, B., Ernst, W., Kunert, R., & Mattanovich, D. (2014). Pichia pastoris secretes recombinant proteins less efficiently than Chinese hamster ovary cells but allows higher space-time yields for less complex proteins. Biotechnology Journal, 9, 526–537.
Werten, M. W. T., Wisselink, W. H., Jansen-van den Bosch, T. J., de Bruin, E. C., et al. (2001). Secreted production of a custom-designed, highly hydrophilic gelatin in Pichia pastoris. Protein Engineering, 14, 447–454.
Trinh, L. B., Phue, J. N., & Shiloach, J. (2003). Effect of methanol feeding strategies on production and yield of recombinant mouse endostatin from Pichia pastoris. Biotechnology and Bioengineering, 82, 438–444.
Fickers, P. (2014). Pichia pastoris: a workhorse for recombinant protein production. Current research in Microbiology and Biotechnology, 2, 354–363.
Saeedinia, A., Shamsara, M., Bahrami, A., Zeinoddini, M., et al. (2008). Heterologous expression of human granulocyte colony stimulating factor in Pichia pastoris. Biotechnology, 2, 1–5.
Bahrami, A., Shojaosadati, S. A., Khalilzadeh, R., Mohammadian, J., Farahani, E. V., & Masoumian, M. R. (2009). Prevention of human granulocyte colony-stimulating factor protein aggregation in recombinant Pichia pastoris fed-batch fermentation using additives. Biotechnology and Applied Biochemistry, 52, 141–148.
Lasnik, M. A., Porekar, V. G., & Stalc', A. (2001). Human granulocyte colony stimulating factor (hG-CSF) expressed by methylotrophic yeast Pichia pastoris. Pfliigers Archives of European Journal of Physiology, 442, R184–R186.
Apte-Deshpande, A., Somani, S., Mandal, G., Soorapaneni, S., & Padmanabhan, S. (2009). Over expression and analysis of O-glycosylated recombinant human granulocyte colony stimulating factor in Pichia pastoris using Agilent 2100 Bioanalyzer. Journal of Biotechnology, 143, 44–50.
Angov, E. (2011). Codon usage: nature’s roadmap to expression and folding of protein. Biotechnology Journal, 6, 650–659.
Macauley-Patrick, S., Fazenda, M. L., McNeil, B., & Harvey, L. M. (2005). Heterologous protein production using the Pichia pastoris expression system. Yeast, 22, 249–270.
Pal, Y., Khusboo, A., & Mukherjee, K. J. (2006). Process optimization of constitutive human granulocyte macrophage colony-stimulating factor (hGM-CSF) expression in Pichia pastoris fed batch culture. Applied Microbiology and Biotechnology, 69, 650–657.
Vogl, T., & Glieder, A. (2013). Regulation of Pichia pastoris promoters and its consequences for protein production. Nature Biotechnology, 30, 385–404.
Viader-Salvado, J. M., Cab-Barrera, E. L., Galan-Wong, L. J., & Guerrero-Olazaran, M. (2006). Genotyping of recombinant Pichia pastoris strains. Cellular and Molecular Biology Letters, 11, 348–359.
Jafari, R., Sundstrom, B. E., & Holm, P. (2011). Optimization of production of the anti keratin 8 single chain Fv TS-218 in Pichia pastoris using design of experiments. Microbial Cell Factories, 10, 34.
Jahic, M., Wallberg, F., Bollock, M., Garcia, P., & Enfors, S. O. (2003). Temperature limited fed-batch technique for control of proteolysis in Pichia pastoris bioreactor cultures. Microbial Cell Factories, 2, 6.
André, N., Cherouati, N., Prual, C., Steffan, T., Zeder-Lutz, G., Magnin, T., Pattus, F., Michel, H., Wagner, R., & Reinhart, C. (2006). Enhancing functional production of G protein-coupled receptors in Pichia pastoris to levels required for structural studies via a single expression system. Protein Science, 15, 1115–1126.
Batra, J., Beri, D., & Mishra, S. (2014). Response surface methodology based optimization of β–glucosidase production from Pichia pastoris. Applied Biochemistry and Biotechnology, 172, 380–393.
Athmaram, T. N., Singh, A. K., Saraswat, S., Srivastava, S., Miisra, P., Rao, M. K., Gopalan, N., & Rao, P. V. L. (2013). A simple Pichia pastoris fermentation and downstream processing strategy for making recombinant pandemic swine origin influenza A virus hemagglutinin protein. Journal of Industrial Microbiology and Biotechnology, 40, 245–255.
Brierley, R. A., Davis, G. R., Holtz, G.. C., Gleeson, M. A., Howard, B. D. (1997) Production of insulin-like growth factor-1 in methylotropic yeast cells. Google Patents, US 08/308, 196.
Werten, M. W. T., Wisselink, W. H., Jansen-van Den Bosch, T. J. C., de Bruin, E. A., & de Wolf, F. (2001). Secreted production of a custom-designed, highly hydrophilic gelatin in Pichia pastoris. Protein Engineering, 14, 447–454.
Ricci, M. S., Sarkar, C. A., Fallon, E. M., Lauffenburger, D. A., & Brems, D. N. (2003). pH dependence of structural stability of interleukin-2 and granulocyte colony-stimulating factor. Protein Science, 12, 1030–38.
Gorgens, J. F., van Zyl, W. H., Knoetze, J. H., & Hahn-Hagerdal, B. (2000). Amino acid supplementation improves heterologous protein production by Saccharomyces cerevisiae in defined medium. Applied Microbiology and Biotechnology, 67, 684–691.
Heyland, J., Fu, J., Blank, L. M., & Schmid, A. (2011). Carbon metabolism limits recombinant protein production in Pichia pastoris. Biotechnology and Bioengineering, 108, 1942–1953.
Ramon, R., Ferrer, P., & Valero, F. (2007). Sorbitol co-feeding reduces metabolic burden caused by the overexpression of Rhizopus oryzae lipase in Pichia pastoris. Journal of Biotechnology, 130, 39–46.
Cos, O., Ramon, R., Montesinos, J. L., & Valero, F. (2006). Operational strategies, monitoring and control of heterologous protein production in the methylotrophic yeast Pichia pastoris under different promoters: a review. Microbial Cell Factories, 5, 17.
Maurer, M., Kuhleitner, M., Gasser, B., & Mattanovich, D. (2006). Versatile modeling and optimization of fed batch processes for the production of secreted heterologous proteins with Pichia pastoris. Microbial Cell Factories, 5, 37.
Werner, R. J. (2004). Economic aspects of commercial manufacture of biopharmaceuticals. Journal of Biotechnology, 113, 171–182.
Chien, S.-F. (2010). Cloning and expression of bioactive human granulocyte colony stimulating factor in Pichia pastoris. Journal of Chinese Chemical Society, 57, 850–856.
Acknowledgments
The authors wish to thank IIT Delhi for providing funds to carry out this research under the “High impact research and technology leadership project.”
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Maity, N., Thawani, A., Sharma, A. et al. Expression and Control of Codon-Optimized Granulocyte Colony-Stimulating Factor in Pichia pastoris . Appl Biochem Biotechnol 178, 159–172 (2016). https://doi.org/10.1007/s12010-015-1865-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-015-1865-y