
Abstract
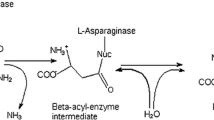
l-Asparaginase (3.5.1.1) is an enzyme widely used to treat the acute lymphoblastic leukemia. Two genes coding for l-asparaginase (ansA1 and ansA3) from Bacillus licheniformis MTCC 429 were cloned and overexpressed in Escherichia coli BL21 (DE3) cells. The recombinant proteins were purified to homogeneity by one-step purification process and further characterized for various biochemical parameters. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis showed that both the enzymes are monomers of ∼37 kDa. Recombinant ansA1 was found to be highly unstable, and recombinant ansA3 was catalytically active and stable, which showed an optimum activity of 407.65 IU/mg at 37 °C and pH 8. Recombinant ansA3 showed higher substrate specificity for l-asparagine with negligible glutaminase activity. Kinetic parameters like K m , V max, k cat, and k cat/K m were calculated for recombinant ansA3.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute lymphoblastic leukemia (ALL) is a case of leukemia affecting mainly children. The therapeutic enzyme l-asparaginase (EC 3.5.1.1) is primarily applied for the treatment of ALL. Asparaginase enzyme is produced by a wide range of organisms which include animals, plants, microbes, and in the serum of certain rodents but not in human beings [1]. The properties of l-asparaginase enzyme have been studied intensively in microorganisms such as Escherichia coli [2, 3], Erwinia carotovora [4], and Erwinia chrysanthemi [5]. Very few drugs are available in the market for the treatment of this disease, and a few more drugs are under clinical trials. These drugs show harsh side effects due to the accompanying glutaminase activity. Glutaminase activity can cause liver dysfunction, pancreatitis, leucopenia, neurological seizures, and coagulation abnormalities leading to intracranial thrombosis or hemorrhage [6]. These side effects are due to the combination of glutaminase activity to the l-asparaginase enzyme. So, efforts are to reduce or eliminate the glutaminase activity from l-asparaginases. Simultaneously, researchers are searching for new microbial sources of l-asparaginase showing reduced or no glutaminase activity. There are reports of l-asparaginase with low glutaminase activity from Bacillus licheniformis [7]. Few studies have reported the use of microbial asparaginase in food industry. Acrylamide formation in heated foods is mainly due to the reaction of free asparagine and reducing sugars. Deamination of asparagine prevents acrylamide formation [8]. Thus, the enzyme can also be applied to thin out the formation of acrylamide in fried and oven-cooked foods, especially in potato chips [9, 10]. Also, there are reports of enzymatic acrylamide mitigation in relation to its use in the food industry [11]. Although production and purification techniques have been developed for l-asparaginase recovery from the bacteria, enzyme yields have been low [12]. To overcome recovery problems, it is extremely desirable to clone, overexpress, and purify the l-asparaginase gene from a microbial source. In the present study, two genes of asparaginase from B. licheniformis MTCC 429 were cloned, overexpressed, and purified. The purified enzyme was further characterized for various parameters. We further demonstrate that one of the two l-asparaginase genes, i.e., ansA1 and ansA3, is catalytically more active than the other.
Materials and Methods
Bacterial Strain and Vector
B. licheniformis MTCC 429 was obtained from Microbial Type Culture Collection (MTCC), Chandigarh, India. The strain was grown and maintained on brain–heart infusion (BHI) media. The culture conditions for growth were 37 °C and pH 7.4. The E. coli DH5α was used as the host for cloning while E. coli BL21 (DE3) was used for cloning, as well as expression of the l-asparaginase genes. The E. coli strains were grown in Luria–Bertani (LB) medium supplemented with ampicillin (100 μg/ml) at 37 °C. The cloning vector used in this study was pRSET A (Invitrogen, USA). Restriction endonucleases were obtained from New England Biolabs (NEB), and T4 DNA ligase was obtained from Invitrogen, USA.
DNA Isolation and Cloning of ansA1 and ansA3 Genes
DNA from B. licheniformis MTCC 429 was isolated using standard procedures [16]. The two genes coding l-asparaginase were amplified with the help of the primer pairs ansA1 FP (5′-GGC CAC AAT GGA TCC ATG AAT AAA AAA GTA GCT CTC-3′) and ansA1 RP (5′-GTA CGC AGC CTC GAG CTA ATA GCA GAA TTT GTC TTT-3′) for ansA1 gene and ansA3 FP (5′-GAA CTC TGG GGA TCC ATG AAA AAG TTA CTG CTG TTG-3′) and ansA3 RP (5′-AGG TTC CAA GAA TTC TTA TAT GAT GAT ATC GTC TGC-3′) for ansA3 gene. The primers were designed to get full-length genes. The restriction sites for forward and reverse primers were, namely, BamHI and EcoRI in ansA1 primers, and BamHI and XhoI were introduced in the ansA3 primers, respectively. The PCR reaction used to amplify the genes consisted of 1.3 μl of 10× Taq buffer, 1.0 μl of forward primer, 1.0 μl of reverse primer, 1.0 μl of dNTPs, 1.0 μl of DNA, 0.35 μl of Taq polymerase, and 6.85 μl of Milli-Q. The final volume of the reaction mixture was 12.5 μl. Amplification is composed of 30 cycles of initial denaturation at 94 °C for 3 min, denaturation for 1 min, followed by annealing at 59 °C for 30 s and extension for 45 s at 72 °C. The final extension was carried out at 72 °C for 3 min. The amplified fragments so obtained were cloned into expression vector pRSET A. With the help of standardized molecular techniques, the plasmid was transformed into E. coli BL21 (DE3) cells.
Overexpression and Purification
ansA1 and ansA3 were expressed in E. coli BL21 (DE3) cells by growing them overnight at 37 °C in LB medium with 100 μg/ml ampicillin. The protein expression was induced using 0.5 mM IPTG. The cells thus grown were harvested with the help of centrifugation at 7,800g for 10 min. The pellet obtained was suspended in sodium phosphate buffer containing NaCl 0.3 M (pH 7.4). Cells were lysed with the help of ultrasonicator (Vibra cell, Sonics, USA) keeping its pulse 30 s on and 30 s off for 5 min. The crude enzyme solution was purified in a single step using affinity chromatography (Ni-NTA, Nucleo-pore, Genetix, India). The column was equilibrated with equilibration buffer potassium phosphate buffer pH 7.4, washed, and protein was eluted as per the user’s guide. The proteins were monitored by measuring protein concentration of the collected fractions with the help of Bio-Rad (Richmond, CA) protein assay method as described by Bradford [13]. To remove excess salts from the purified proteins, the protein solution was dialyzed at 4 °C against the same buffer (sodium phosphate buffer, pH 7.4) and stored at −20 °C.
Molecular Weight of Purified Recombinant Proteins
Purification of the proteins was checked by polyacrylamide gel electrophoresis (PAGE) analysis. To determine the subunit molecular weight, denatured purified proteins were separated according to their molecular weight on 10 % sodium dodecyl sulfate (SDS)-PAGE 1.5 mm gel [14]. Denaturation of purified proteins was done at 95 °C for 5 min. The protein bands were stained with Coomassie Brilliant Blue R-250 dye. The molecular weight of the purified proteins was determined with the help of the medium range protein molecular weight markers (Merck, India). Native PAGE was performed under nondenaturing conditions.
Determination of Asparaginase Activity
The enzyme activity was determined using Nessler’s reagent [15]. One international unit (IU) of l-asparaginase is the quantity of enzyme which liberates 1 μmol of ammonia in 1 min at 37 °C.
Effect of pH on Asparaginase Activity
Effect of pH on purified asparaginase was determined at various pH 6.0, 7.0, 7.5, 8.0, 9.0, and 10.0. The pH range from 6.0 to 10.0 for enzyme activity was checked at 37 °C. For pH 6.0, 7.0, 7.5, and 8.0 and for pH 9.0 and 10.0, potassium phosphate buffers and glycine-sodium hydroxide buffers were used, respectively.
Effect of Temperature on Asparaginase Activity
Temperature study was carried out at different temperatures like 25, 30, 37, 45, and 50 °C. The temperature at which the enzyme showed maximum asparaginase activity was selected for further studies.
Substrate Specificity
Substrate specificity of the purified enzyme against different substrates such as l-asparagine, d-asparagine, l-aspartic acid, l-glutamine, and l-glutamic acid was determined at 37 °C and pH 8. The activity of the enzyme toward different substrates was determined as described for l-asparaginase activity using Nessler’s method. The activity was measured at 40-mM concentration of each substrate.
Effectors Affecting Asparaginase Activity
Different effectors were used to evaluate the activity of recombinant l-asparaginase at a concentration of 5 mM. The effectors used were EDTA (a metal chelator), Ca2+, Hg2+, Ni2+, Mg2+, Mn2+, Fe3+, Na+, and K+. The activity of recombinant enzyme in the presence of effectors was determined at 37 °C and pH 8.
Determination of Kinetic Parameters
The kinetic parameters of the purified recombinant enzyme were determined using l-asparagine as a substrate at various concentrations from 10 to 300 mM in phosphate buffer (pH 8) at 37 °C. K m and V max were determined by Lineweaver–Burk plot according to Michaelis and Menten equation. The parameters like k cat (turnover number) and k cat/K m were calculated on the basis of one active site per 37 kDa subunit molecular mass.
Results and Discussion
DNA Isolation and Amplification of ansA1 and ansA3 Genes
Good quality genomic DNA was isolated [16], and both ansA1 and ansA3 (accession number KC854793) genes were amplified by PCR. Clear bands of both the genes showing a size of 1 kb were observed under UV transilluminator after agarose gel electrophoresis (Fig. 1).

PCR amplification of both ansA1 and ansA3 genes: step-up 100-bp marker (lane A), amplification of ansA1 gene (lane B), and amplification of ansA3 gene (lane C)
Overexpression and Purification
The overexpressed recombinant proteins (rBliAI and rBliAIII) of B. licheniformis MTCC 429 were purified to homogeneity by affinity chromatography using Ni-NTA column. Overexpression of recombinant His-tagged asparaginase into E. coli leads to improvement in yield and affinity purification process of enzyme [17]. The fractions showing presence of protein with the help of Bradford’s reagent were pooled together. The pooled protein solution was dialyzed against the same buffer and was checked for the l-asparaginase activity.
The purified protein after PAGE analysis showed a single band (Fig. 2) illustrating its homogeneous nature. The molecular weight of the subunits of ansA1 and ansA3 was found to be approximately 37 kDa after SDS-PAGE analysis. SDS-PAGE analysis of recombinant l-asparaginase from Pyrococcus furiosus displayed single 37-kDa band [18]. In another study, recombinant ansA from Rhizobium etli in SDS-PAGE showed the presence of a single polypeptide chain of 47 kDa [17]. Native PAGE analysis showed the molecular weight of the purified protein as ∼74 kDa. This study confirmed that the ansA1 and ansA3 enzymes from B. licheniformis MTCC 429 are a homodimer in nature with two subunits of 37 kDa (Fig. 2). Some studies showed that asparaginase II of E. coli is an active tetramer with a single subunit of 35.6 kDa [19].

PAGE analysis: nondenatured sample (a), medium range molecular weight marker (b), purified rBliAI (c), and purified rBliAIII (d)
Characterization of Purified rBliAI and rBliAIII
There are reports of two differentially regulated genes encoding l-asparaginase from Bacillus subtilis 168 [20]. Similarly, while carrying out the characterization of both recombinant (rBliAI and rBliAIII) enzymes from B. licheniformis MTCC 429 when expressed in E. coli (DE3), it was observed that recombinant ansA1 enzyme was highly unstable and showed very low activity as compared to recombinant ansA3 enzyme. In earlier reports, E. coli type II l-asparaginase (EcA-II) was found to have a higher affinity for l-asparagine than the type I isozyme (EcA-I) [21]. Therefore, recombinant ansA3 enzyme was selected for further studies. The results of the purified rBliAIII are summarized in Table 1.
Effect of pH
The purified recombinant enzyme was active in a pH range of 7.0–9.0. Maximum asparaginase activity was found at pH 8.0. The enzyme was inactivated at highly acidic and alkaline conditions. The effect of pH on activity is shown in Fig. 3, wherein it can be determined that the enzyme exhibited maximum activity at pH 8. Youssef and Omair [22] showed that purified l-asparaginase II from E. coli W3110 was active in alkaline solutions. At ranges above or below pH 8, the enzyme showed less activity, but at very low or very high pH, the enzyme activity was absent. A similar pH value was reported for l-asparaginase enzyme from different Streptomyces sp. at pH 8 [23–25].

Effect of pH on purified recombinant enzyme activity
Effect of Temperature
It has been demonstrated in the studies that temperature is the most important variable that modifies the enzymatic activity of l-asparaginase [17]. The reaction rate of l-asparaginase was measured at various temperatures; maximum activity was obtained at 37 °C. At higher temperatures, the reaction rate declined sharply (Fig. 4). Maximum activity of l-asparaginase from Pseudomonas aeruginosa 50071 [1], E. carotovora [26], and Cladosporium sp. [27] have been observed at 37, 35, and 30 °C, respectively.

Effect of temperature on purified recombinant enzyme activity
Substrate Specificity
The substrate specificity of the recombinant enzyme, l-asparaginase enzyme, activity was determined at a 40 mM concentration of different substrate analogs like d-asparagine, l-glutamine, l-aspartic acid, and l-glutamic acid. Enzyme activity was not detected with d-asparagine monohydrate and l-glutamic acid as substrates, whereas very less activity was observed with l-glutamine and l-aspartic acid. The relative activity was expressed as percentage activity. The activity of recombinant l-asparaginase was highly specific for l-asparagine (Fig. 5). This attribute makes the recombinant l-asparaginase potentially very useful in the treatment of ALL, as the side effects in l-asparaginase therapy are mainly due to the contamination by glutaminase of l-asparaginase [28]. Purified enzyme from E. carotovora exhibited a low glutaminase activity, which was nearly 1.5 % of that of l-asparaginase activity which was much lower than that exhibited by the E. coli and E. chrysanthemi enzymes (∼10 %) [4]. In the present study, the purified recombinant ansA3 exhibited very negligible glutaminase activity of 0.6 % of that of l-asparaginase which is quite lower than reported in earlier studies. l-Asparaginase having reduced glutaminase activity is better for ALL treatment [29].

Substrate specificity of recombinant enzyme
Effect of Various Metal Ions and EDTA on Enzyme Activity
Metal ions play important role in the biological functions of many enzymes [30]. In order to evaluate the effects of some metal ions on the enzymatic activity, enzyme reaction in the presence of l-asparagine along with metal ions was carried out. Effect of various metal ions at a concentration of 5 mM on enzyme activity is shown in Fig. 6. In the presence of Mg2+, Mn2+, and Na+, it was observed that there was an increase in the recombinant ansA3 activity, whereas complete loss in activity was observed in the presence of HgCl2. Reduced enzyme activity was noted in the presence of Ni2+, Ca2+, K+, urea, and Fe3+. Similar observations were made in the presence of a metal chelator, EDTA.

Effect of effectors (5 mM) on enzyme activity
Kinetic Parameters
The K m and V max values were obtained with the help of Lineweaver–Burk plot (Fig. 7). l-Asparagine was hydrolyzed by the enzyme with a Michaelis and Menten constant (K m ) of 0.671 × 10−3 M, and V max was found to be 36.60 μmol/min. The K m value of recombinant ansA3 from B. licheniformis MTCC 429 indicated that the enzyme has higher affinity toward l-asparagine compared to l-asparaginase from Erwinia aroideae [31, 32], Corynebacterium glutamicum [33], Cylindrocarpon obtusisporum [34], Mycobacterium phlei [35], Thermus thermophilus [36], Tetrahymena pyriformis [37], and B. subtilis [38]. The K m values of l-asparaginases from different microbial sources are shown in Table 2.

Lineweaver–Burk plot
The kinetic properties of recombinant BliAIII were investigated, and the parameters like k cat and k cat/K m were also studied with l-asparagine as a substrate. Comparative studies of the rBliAIII with asparaginases from various microbial sources showed that rBliAIII has lower K m value for l-asparagine (Table 2), confirming that rBliAIII has better affinity toward its substrate than other reported microbial sources. The catalytic constant (k cat) of rBliAIII was only 1.5 times higher than the l-asparaginase from E. coli, and catalytic constant of rErAII was 1.35 times higher than rBliA (Table 3). The absolute value of k cat/K m suggests the catalytic efficiency of the enzyme [39]. k cat/K m of the rBliAIII was 5.4 × 104 which is lower than the asparaginases from E. coli and Erwinia.
Conclusion
Cloning, expression, and purification of recombinant ansA1 and ansA3 showed that ansA3 is a potential enzyme which could be used in the treatment of ALL as recombinant ansA3 showed higher substrate affinity toward l-asparagine, when the enzyme activity was assessed with different substrate analogs. The enzyme also showed negligible glutaminase activity which further makes it ideal for its employment in pharmaceutical application. The K m value of rBliAIII is 0.67 × 10−3 M which showed that it has a good substrate affinity toward l-asparagine. rBliAIII has a good affinity toward the substrate when compared to various microbial asparaginases. rBliAIII can serve as a potential substitute for the commercially available asparaginase sources. Apart from its application in the pharmaceutical industry, asparaginase enzyme can also be engineered for its application in the food industry to reduce acrylamide formation in starch-based foods.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- ansA1:
-
l-Asparaginase I
- ansA3:
-
l-Asparaginase III
- rBliAI:
-
Recombinant l-asparaginase I from Bacillus licheniformis
- rBliAIII:
-
Recombinant l-asparaginase III from Bacillus licheniformis
- PAGE:
-
Polyacryamide gel electrophoresis
References
El-Bessoumy, A. A., Sarhan, M., & Mansour, J. (2004). Production, isolation, and purification of L-asparaginase from Pseudomonas aeruginosa 50071 using solid-state fermentation. Journal of Biochemistry and Molecular Biology, 37, 387–393.
Bagert, U., & Röhm, K. H. (1989). On the role of histidine and tyrosine residues in E. coli asparaginase; chemical modification and 1H nuclear magnetic resonance studies. Biochimica et Biophysica Acta, 999, 36–41.
Harms, E., Wehner, A., Jennings, M. P., Pugh, K. J., Beacham, I. R., & Roehm, K. H. (1991). Construction of expression systems for Escherichia coli asparaginase II and two-step purification of the recombinant enzyme from periplasmic extracts. Protein Expression and Purification, 2, 144–150.
Kotzia, G. A., & Labrou, N. E. (2005). Cloning, expression and characterisation of Erwinia carotovora L-asparaginase. Journal of Biotechnology, 119, 309–323.
Aghaiypour, K., Wlodawer, A., & Lubkowski, J. (2001). Structural basis for the activity and substrate specificity of Erwinia chrysanthemi L-asparaginase. Biochemistry, 40, 5655–5664.
Duval, M., Suciu, S., Ferster, A., Rialland, X., Nelken, B., Lutz, P., Benoit, Y., Robert, A., Manel, A. M., Vilmer, E., Otten, J., & Philippe, N. (2002). Comparison of Escherichia coli-asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment of Cancer-Children’s Leukemia Group phase 3 trial. Blood, 99, 2734–2739.
Mahajan, R. V., Saran, S., Kameswaran, K., Kumar, V., & Saxena, R. K. (2012). Efficient production of L-asparaginase from Bacillus licheniformis with low-glutaminase activity: optimization, scale up and acrylamide degradation studies. Bioresource Technology, 125, 11–16.
Ciesarova, Z., Kiss, E., & Boegl, P. (2006). Impact of L-asparaginase on acrylamide content in potato products. Journal of Food and Nutrition, 45, 141–146.
Rosen, J., & Hellenas, K. E. (2002). Analysis of acrylamide in cooked foods by liquid chromatography tandem mass spectrometry. Analyst, 127, 880–882.
Tareke, E., Rydberg, P., Karlsson, P., Eriksson, S., & Tornqvist, M. (2002). Analysis of acrylamide, a carcinogen formed in heated food stuffs. Journal of Agricultural and Food Chemistry, 50, 4998–5006.
Pedreschi, F., Kaack, K., & Granby, K. (2008). The effects of asparaginase on arylamide formation in French fries. Food Chemistry, 109, 386–392.
Kenari, S. L. D., Alemzadeh, I., & Maghsodi, V. (2011). Production of L-asparaginase from Escherichia coli ATCC 11303: optimization by response surface methodology. Food and Bioproducts Processing, 89, 315–321.
Bradford, M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685.
Imada, A., Igarasi, S., Nakahama, K., & Isono, M. (1973). Asparaginase and glutaminase activities of micro-organisms. Journal of General Microbiology, 76, 85–99.
Sambrook, J., Russell, D. W., & Cold Spring Harbor Laboratory. (2001). Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory.
Moreno-Enriquez, A., Evangelista-Martinez, Z., Gonzalez-Mondragon, E. G., Calderon-Flores, A., Arreguin, R., Perez-Rueda, E., & Huerta-Saquero, A. (2012). Biochemical characterization of recombinant L-asparaginase (AnsA) from Rhizobium etli, a member of an increasing rhizobial-type family of L-asparaginases. Journal of Microbiology and Biotechnology, 22, 292–300.
Bansal, S., Gnaneswari, D., Mishra, P., & Kundu, B. (2010). Structural stability and functional analysis of L-asparaginase from Pyrococcus furiosus. Biochemistry (Mosc), 75, 375–381.
Epp, O., Steigemanhne, W., & Formanekan Huber, R. D. (1971). Crystallographic evidence for the tetrameric subunit structure of L-asparaginase from Escherichia coli. European Journal of Biochemistry, 20, 432–437.
Fisher, S. H., & Wray, L. V. (2002). Bacillus subtilis 168 contains two differentially regulated genes encoding L-asparaginase. Journal of Bacteriology, 184, 2148–2154.
Cedar, H., & Schwartz, J. H. (1967). Localization of the two-L-asparaginases in anaerobically grown Escherichia coli. Journal of Biological Chemistry, 242, 3753–3755.
Youssef, M. M., & Al-Omair, M. A. (2008). Cloning, purification, characterization and immobilization of L-asparaginase II from E. coli W3110. Asian Journal of Biochemistry, 3, 337–350.
Basha, S. N., Rekha, R., Komala, M., & Ruby, S. (2009). Production of extracellular anti-leukemic enzyme L-asparaginase from marine actinomycetes by solid state and submerged fermentation: purification and characterization. Tropical Journal of Pharmaceutical Research, 8, 353–360.
Dharmaraj, S. (2011). Study of L-asparaginase production by Streptomyces noursei MTCC 10469, isolated from marine sponge Callyspongia diffusa. Iranian Journal of Biotechnology, 9, 102–108.
Dhevagi, P., & Poorani, E. (2006). Isolation and characterization of L-asparaginase from marine actinomycetes. Indian Journal of Biotechnology, 5, 514–520.
Kamble, V. P., Rao, R. S., Borker, P. S., Khobragade, C. N., & Dawane, B. S. (2006). Purification of L-asparaginase from a bacteria Erwinia carotovora and effect of adihydropyrimidine derivative on some of its kinetic parameters. Indian Journal of Biochemistry and Biophysics, 43, 391–394.
Mohankumar, N. S., & Manonmani, H. K. (2013). Purification, characterization and kinetic properties of extracellular L-asparaginase produced by Cladosporium sp. World Journal of Microbiology and Biotechnology, 29, 577–587.
Manna, S., Sinha, A., Sadhukhan, R., & Chakrabarty, S. L. (1995). Purification, characterization and antitumor activity of L-asparaginase isolated from Pseudomonas stutzeri MB-405. Current Microbiology, 30, 291–298.
Ramya, L. N., Doble, M., Rekha, V. P., & Pulicherla, K. K. (2012). L-Asparaginase as potent anti-leukemic agent and its significance of having reduced glutaminase side activity for better treatment of acute lymphoblastic leukaemia. Applied Biochemistry and Biotechnology, 167, 2144–2159.
Riordan, J. F. (1977). The role of metals in enzyme activity. Annals of Clinical and Laboratory Science, 7, 119–129.
Peterson, R. E., & Ciegler, A. (1969). L-Asparaginase production by Erwinia aroideae. Applied Microbiology, 18, 64–67.
Tiwari, N., & Dua, R. D. (1996). Purification and preliminary characterization of L-asparaginase from Erwinia aroideae NRRL B-138. Indian Journal of Biochemistry and Biophysics, 33, 371–376.
Mesas, J. M., Gil, J. A., & Martin, J. F. (1990). Characterization and partial purification of L-asparaginase from Corynebacterium glutamicum. Journal of General Microbiology, 136, 515–519.
Raha, S. K., Roy, S. K., Dey, S. K., & Chakrabarty, S. L. (1990). Purification and properties of an L-asparaginase from Cylindrocarpon obtusisporum MB-10. Biochemistry, 21, 987–1000.
Pastuszak, I., & Szymona, M. (1975). L-asparaginase activity of Mycobacterium phlei under various growth conditions. Acta Microbiologica Polonica. Series A, 8, 131–139.
Pritsa, A. A., & Kyriakidis, D. A. (2001). L-asparaginase of Thermus thermophilus: purification, properties and identification of essential amino acids for its catalytic activity. Molecular and Cellular Biochemistry, 216, 93–101.
Triantafillou, D. J., Georgatsos, J. G., & Kyriakidis, D. A. (1988). Purification and properties of a membrane-bound L-asparaginase of Tetrahymena pyriformis. Molecular and Cellular Biochemistry, 81, 43–51.
OnishiY, Y. S., Thongsanit, J., Takagi, K., Yoshimune, K., & Wakayama, M. (2011). Expression in Escherichia coli of a gene encoding type II L-asparaginase from Bacillus subtilis, and characterization of its unique properties. Annals of Microbiology, 61, 517–524.
Price, N. C., & Nairn, J. (2009). Exploring proteins: a student’s guide to experimental skills and methods (pp. 104–148). New York: Oxford University Press.
Derst, C., Henseling, J., & Rohm, K. H. (2000). Engineering the substrate specificity of Escherichia coli asparaginase II. Selective reduction of glutaminase activity by amino acid replacements at position 248. Protein Science, 9, 2009–2017.
Kotzia, G. A., & Labrou, N. E. (2007). L-Asparaginase from Erwinia chrysanthemi 3937: cloning, expression and characterization. Journal of Biotechnology, 127, 657–669.
Roth, G., Nunes, J. E. S., Rosado, L. A., Bizarro, C. V., Volpato, G., Nunes, C. P., Renard, G., Basso, L. A., Santos, D. S., & Chies, J. M. (2013). Recombinant Erwinia carotovora L-asparaginase II production in Escherichia coli fed-batch cultures. Brazilian Journal of Chemical Engineering, 30, 245–256.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sudhir, A.P., Dave, B.R., Prajapati, A.S. et al. Characterization of a Recombinant Glutaminase-Free l-Asparaginase (ansA3) Enzyme with High Catalytic Activity from Bacillus licheniformis . Appl Biochem Biotechnol 174, 2504–2515 (2014). https://doi.org/10.1007/s12010-014-1200-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-014-1200-z