
Abstract
l-asparaginases (l-ASNases; EC 3.5.1.1) are aminohydrolases that catalyze the hydrolysis of l-asparagine (l-Asn) to l-aspartic acid and ammonia, resulting in the death of acute lymphoblastic leukemic cells and other blood cancer cells. In this study, Bacillus sonorensis (accession number MK523484) uncharacterized l-ASNase gene (accession number MN562875) was isolated by polymerase chain reaction (PCR), cloned into pET28a (+) vector, and expressed in Escherichia coli as a cytosolic protein. The recombinant enzyme was purified by affinity chromatography at 23.79-fold and 49.37% recovery. Denaturing polyacrylamide gel (10%) analysis of the purified enzyme resulted in a single protein band at 36 kDa that immunoreacted strongly with 6His-tag monoclonal antibody. The purified enzyme exhibited optimal activity at 45 °C and pH 7.0 and retained 92% and 85% of its initial activity after incubation for 60 min at 37 °C and 45 °C, respectively. The purified enzyme exhibited substrate specificity toward l-asparagine and low glutaminase activity (15.72%) toward l-glutamine at a concentration of 10 mM. The Km and Vmax values were 2.004 mM and 3723 µmol min1−, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Microbial l-asparaginases (l-ASNases), which were described five decades earlier, have been found to be effective drug proteins in the treatment of various oncological pathologies, ranging from hematologic cancer such as pediatric acute lymphoblastic leukemia (ALL), acute myeloblastic leukemia, and lymphoma (lymph sarcoma) to other high-mortality malignancies [1]. Microbial l-ASNases are aminohydrolases with narrow substrate specificities; they act through the hydrolysis of l-asparagine (and l-glutamine) to l-aspartic (l-Asp) and l-glutamic (l-Glu) acids with the concomitant release of ammonia, resulting in the depletion of the two vital amino acids l-Asn and l-Gln required for the synthesis of nucleic acids and proteins in the blood circulation of patients with ALL and death of leukemic cells rather than normal cells [2, 3]. Unlike normal cells, leukemic cells cannot synthesize l-Asn de novo due to either low expression level of asparagine synthetase (ASNS) gene or complete absence of this gene; hence these cells require an external source of l-Asn for survival [4,5,6,7]. Microbial l-ASNases are generally available in three forms till date, viz., native l-ASNase from Escherichia coli and Dickeya chrysanthemi (formerly named Erwinia chrysanthemi) and PEGylated or polyethylene glycol-conjugated E. coli L-ASNase (PEG-asparaginase). Both native and PEGylated E. coli L-ASNases are used as the first-line treatment option for ALL, whereas native D. chrysanthemi L-ASNase can be applied as the second-line treatment [8]. One of the major differences between these commercial l-ASNase preparations is the blood circulation elimination half-life, because the PEGylated l-ASNase form has five times longer half-life than native E. coli l-ASNase and nine times longer half-life than native D. chrysanthemi l-ASNase. Furthermore, D. chrysanthemi l-ASNase has a similar l-asparaginase activity to that of E. coli l-ASNase but 10 times higher glutaminase activity [9,10,11]. However, the glutaminase activity of microbial l-ASNases is debatable. Recently, it has been demonstrated that the accompanied glutaminase activity of l-ASNase is required for antileukemic and anticancer effects, especially in patients with ALL positive for ASNS, but it is not essential for ASNS-negative childhood ALL [9,10,11,12,13,14]. Despite successful use of l-ASNase drug for oncolytic remission, FDA-approved prokaryotic l-ASNase could induce some problems, due to which its safety profile in humans remains an obstacle [15]. One of such issues is the hypersensitivity reaction manifested in some patients with ALL treated with l-ASNase due to the production of antibodies in response to l-ASNase. Therefore, rapid inactivation of the injected enzyme (silent hypersensitivity or silent inactivation) and worse prognosis for this reason continuous monitoring of l-ASNase activity in patient’s blood is important [16, 17]. l-asparaginase along with glutaminase coactivity depletes both plasma glutamine and asparagine levels, resulting in protein synthesis disruption and hyperammonemia due to the deamination reaction, which contribute to hypersensitivity and neurotoxicity symptoms observed in patients with ALL [18].
In the present research, we isolated uncharacterized l-ASNase gene from Bacillus sonorensis for the first time, followed by cloning and expression in E. coli and purification and characterization of the enzyme. This study was aimed to develop a potential alternative therapeutic protein for ALL overcoming as possible side effects in currently used and approved chemotherapeutic regimens.
2 Materials and Methods
2.1 Bacterial Strains and Cloning Vectors
B. sonorensis strain, a source of l-ASNase gene, was isolated from soil garden in Alexandria, Egypt, and identified using biochemical tests and 16S rRNA gene sequencing method (GenBank database accession number MK523484). E. coli BL21 (DE3) pLysS strain was used as the expression host, and pGEM®-T Easy (Promega, USA) and pET28a (+) (Novagen, USA) were used as vectors.
2.2 l-ASNase Gene Isolation and Cloning into pGEM-T-Easy Vector
The genome of B. sonorensis (https://www.ncbi.nlm.nih.gov/nucleotide/NZ-CP021920.1) revealed an ORF of 933 bp encoding for l-ASNase gene that exhibited homology with other Bacillus sp. l-ASNases. A set of primers was designed to isolate the l-ASNase gene, with the sequence forward (5′-GGATCCATGAAAAAATTACTGCTGTTAACC-3′) and reverse (5′-GAATTCAATGATGATATCGTCTGCAATCGG-3′). The underlined sequences are EcoRI and BamHI restriction sites. DNA was isolated from B. sonorensis strain using Wizard® Genomic DNA Purification Kit (Promega, USA) according to the instruction manual, and 100 ng of DNA was used to amplify the l-ASNase gene. The PCR product of the l-ASNase gene was first ligated onto the pGEM-T-Easy™ plasmid, and then the recombinant plasmid was used to transform competent E. coli JM109 cells, as described by Sambrook et al. [19]. Plasmids were isolated from some recombinant clones, and sequencing was conducted as described by Sanger et al. [20] using the multiple cloning site universal primers T7 and SP6. Nucleotide sequences were analyzed using BLASTn at https://www.ncbi.nlm.nih.gov/, using BioEdit and Jalview programs.
2.3 Subcloning of B. sonorensis l-ASNase Gene into pET28a (+) Plasmid and Expression in E. coli
The recombinant pGEM-T-Easy™ vector carrying the B. sonorensis l-ASNase gene was digested with EcoRI and BamHI as described by Sambrook et al. [19]. The product of the digestion reaction was resolved by 1% agarose gel electrophoresis, and the l-ASNase gene insert purified from the agarose gel was cloned into the ready-to-use pET28a (+) vector. The recombinant pET28a (+) vector carrying the B. sonorensis l-ASNase gene was transformed into E. coli BL21 (DE3) pLysS competent cells. Next, the cells were cultured at 37 °C for 16 h with agitation at 200 rpm in LB medium containing lactose as an inducer at a concentration of 2 g/L and 34 µg/mL kanamycin. After incubation, cells were collected by centrifugation at 8000 rpm for 20 min at 4 °C, resuspended thoroughly in 50 mM potassium phosphate buffer (pH 7.5) containing 10% glycerol, and sonicated using 4 × 15-s pulses. The supernatant containing the recombinant l-ASNase was collected by centrifugation at 12,000 rpm for 10 min at 4 °C.
2.4 Quantification of Protein
Total cell proteins and purified recombinant enzyme were determined according to the protocol described by Bradford [21]. A standard curve was generated using stock BSA at 0.5 µg/mL concentration, and serial dilutions were made to generate the standard curve starting from 2.5 to 50 µg BSA.
2.5 Measurement of Enzyme Activity
The l-ASNase activity was determined by measuring the quantity of the released ammonium from l-Asn at 450 nm using Nessler’s reagent [22]. One micromole of ammonia released per minute is equal to 1 unit (1 U) of the recombinant enzyme, as assessed using the ammonium chloride standard curve, at a concentration of 1 mM.
2.6 SDS-PAGE and Western Blotting Analysis
The purity and molecular weight of the protein were analyzed by 10% SDS-PAGE according to Laemmli protocol [23]. The recombinant B. sonorensis l-ASNase was validated by western blotting using monoclonal antibody against the 6His-tag fusion peptide at 1:1000 dilution as described by Towbin et al. [24]. Goat anti-mouse horseradish peroxidase–conjugated IgG was used as the secondary antibody at 1:2000 dilution, and the localization of the recombinant l-ASNase on the nitrocellulose membrane was detected using the 3, 3′, 5, 5′-tetramethylbenzidine (TMB) liquid substrate detection system.
2.7 Recombinant Protein Purification using Affinity Resin
The 6His-tag fusion recombinant l-ASNase was purified using nickel affinity resin. The affinity resin was poured into a 3 × 1 cm column and washed thoroughly with 5-bed volumes of deionized water, followed by 5-bed volumes of equilibration buffer consisting of 20 mM potassium phosphate buffer (pH 7.5) and imidazole at 20 mM concentration [25]. Total protein extract was loaded into the column, followed by washing with the equilibration buffer till the absorbance at 280 nm reached zero, after which the bound recombinant enzyme was eluted using 500 mM imidazole in the equilibration buffer. Fractions representing the second chromatographic peak that contained the recombinant l-ASNase were pooled together and dialyzed overnight against 20 mM potassium phosphate buffer (pH 7.5).
2.8 Sequence Analysis, Construction of Phylogenetic Tree, and Structural Modeling
Sequence analysis of the B. sonorensis l-ASNase gene (933 bp) was performed using BLASTn and BLASTp available at NCBI, and multiple sequence alignment was conducted using the JalView 2.10.5 program. Phylogenetic tree construction and sequence annotation by structure analysis were conducted as described by Dereeper et al. [26] and Milburn et al. [27]. The amino acid sequence of B. sonorensis l-ASNase was submitted to the Swiss-Model server to predict the 3D structure of the protein, after which the PDB viewer program was used to analyze the structural data. Fetching for the l-ASNase 3D structure template was then performed by BLAST and HHBlits searching against the Swiss-Model template library, and finally, the highest-quality template was selected to construct the l-ASNase 3D model [28, 29].
2.9 Biochemical Characterization of Recombinant B. sonorensis l-ASNase
2.9.1 Optimum Temperature and Thermostability
The optimum temperature was determined by analyzing the l-ASNase activity at different incubation temperatures, ranging from 25 to 60 °C. Thermostability was examined by incubating the purified enzyme at temperatures ranging from 28 to 50 °C for 1 h, and after 10-min time intervals, the percentage residual activity was measured as described previously [30].
2.9.2 Optimum pH
The optimum pH of the purified recombinant B. sonorensis l-ASNase was determined by measuring the enzyme activity at different pH values, ranging from 4 to 10, as described by Saeed et al. [30].
2.9.3 Specificity of Recombinant l-ASNase toward Reaction Substrates
The specificity toward the reaction substrate was analyzed using l-Asn, l-Gln, urea, and acrylamide at a concentration of 10 mM.
2.9.4 Determination of Km and Vmax
The kinetic parameters of B. sonorensis l-ASNase Km and Vmax were determined using different concentrations of l-Asn substrate by the Hyper 32 software.
2.9.5 Effect of Some Compounds on l-ASNase Activity
The effects of metal ions (chloride and sulfate forms), chelating agents (EDTA), and reducing agents (β-mercaptoethanol and DTT) on l-ASNase activity were investigated by incubating the purified recombinant l-ASNase with the test compounds at 1 and 5 mM separately for 15 min on ice after the incubation period, and the residual activity was determined. The effect of l-Gly on l-ASNase activity was investigated. The purified recombinant l-ASNase was incubated at 4 °C and 25 °C with l-Gly at 250 and 500 mM for 3 and 48 h, followed by dialysis and determination of residual activity.
3 Results and Discussion
3.1 B. sonorensis l-ASNase Gene Isolation, Sequence Analysis, and Phylogenetic Tree Construction
Searching the GenBank database for B. sonorensis genome (https://www.ncbi.nlm.gov/nucleotide/NZ_CP021920.1) resulted in the presence of uncharacterized l-ASNase full-length gene consisting of 990 nucleotides coding for the ORF of 329 amino acid residues. The coding sequence was used to design a gene-specific primer set to amplify and further clone the full-length l-ASNase gene (Fig. 1). The amplified 933-bp PCR product was cloned into the pGEM-T-Easy vector and sequenced using T7 and SP6 primers. BLASTp analysis of the B. sonorensis l-ASNase protein-coding sequence (Fig. 2) revealed significant similarity to another Bacillus sp. l-ASNase in the GenBank database (Table 1). It was observed that the identity percentage score with B. licheniformis (GenBank accession number WP_080623552.1) was as high as 95.14% compared to 32.11%, 25.36%, and 27.22% for E. coli (GenBank accession number WP_154293749.1), D. chrysanthemi (GenBank accession number WP_012770786.1), and Staphylococcus aureus (GenBank accession number KII20890.1), respectively. Alignment of the primary structure of B. sonorensis l-ASNase with that of l-ASNases from another Bacillus species indicated that B. sonorensis l-ASNase had > 95% sequence similarity with other Bacillus l-ASNases (Fig. 3). Phylogenetic tree analysis of B. sonorensis l-ASNase (Fig. 4) demonstrated that although B. sonorensis l-ASNase exhibited sequence similarity with other Bacilli l-ASNases, it switched to different clusters from other species such as E. coli, Erwinia, and Vibrio alginolyticus.

Agarose gel (1%) electrophoresis for PCR products of B. sonorensis l-ASNase gene (Lanes 2–4). Lane 1 represents 100-bp DNA ladder

Nucleotide sequence and primary structure of B. sonorensis recombinant l-ASNase. Important amino acid residues and regions include catalytic residues to act as asparaginase are in box; catalytic residues for l-glutaminase are bold underlined; start codon, ATG (Met) is in bold, and asterisk (*) represents stop codon (TAG)

Multiple sequence alignment of the deduced amino acid sequence of B. sonorensis l-ASNase with those of other species. Alignment was created using the following asparaginase sequences (NCBI accession numbers are in parentheses): Bacillus licheniformis (WP_080623552.1); B. paralicheniformis (WP_145653281.1); Bacillus sp. SB47 (WP_026580320.1); B. haynesii (WP_043926860.1); B. swezeyi (WP_076759715.1); B. glycinifermentans (WP_065894701.1); Bacillus sp. TH008 (WP_046129362.1); Risungbinella massiliensis (WP_044642564.1); B. vallismortis (WP_121642831.1); and B. pumilus (WP_058013330.1). Hydrophobic amino acids are in blue; positively charged amino acids are in red; negatively charged amino acids are in magenta; polar amino acids are in green; cysteines are in pink; glycines are in orange; proline amino acid is in yellow; aromatic amino acids are in cyan; and unconserved amino acids are in white (Color figure online)

Phylogenetic relationship of B. sonorensis l-ASNase and sequences from other species
3.2 B. sonorensis l-ASNase Sequence Analysis and Modeling of 3D Structure
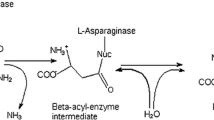
The analysis of B. sonorensis l-ASNase primary structure and elements of the secondary structure (Figs. 2 and 5) disclosed some interesting characteristics. First, there was a common signature for l-ASNases represented by the amino acid residues Thr11,85, Leu22, Ala23, Asp86, and Lys156. These amino acid residues are responsible for the first type of activity characterize most microbial l-ASNase, the l-ASNase activity. Second, B. sonorensis l-ASNase showed the l-glutaminase active site signature represented by the amino acid residues Thr12,85, Leu22, and Glu277 (Table 2). Several microbial l-ASNases such as those of E. coli and Erwinia have dual enzymatic activities toward l-Asn and l-Gln, and accordingly, they can be classified into type I cytosolic enzymes with low glutaminase activity (2–10%) and type II periplasmic l-ASNases with comparable l-asparaginase and l-glutaminase activities [31]. As B. sonorensis l-ASNase is capable of catalyzing both l-Asn and l-Gln, it can be classified into type II l-ASNase; however, the level of l-glutaminase activity required for therapeutic efficacy in the treatment of childhood ALL and other blood cancers still remains highly debated [32,33,34]. Figure 5 shows the components of the secondary structure of l-ASNase. It was observed that B. sonorensis l-ASNase consisted of 9α helices (34%), 17β strands (25.8%), 10.3% turns, and 29.8% coil structure. The isoelectric point pI was predicted to be 5.16. The complete list of amino acid residues involved in the binding of metals and ligands is shown in Table 2. To fetch for related structures and protein models matching the B. sonorensis target l-ASNase sequence, the Swiss-Model template library was searched using BLAST [35] and HHBlits [36], which resulted in 50 templates. Surprisingly, the B. sonorensis l-ASNase Swiss-Model template revealed significant sequence homology and 42.15% identity with Thermococcus kodakarensis l-ASNase, which were centralized and conserved in the secondary structure components of the protein, i.e., the α helices and the β sheets. Figure 6b shows that the predicted 3D structure of B. sonorensis l-ASNase was similar to that of T. kodakarensis l-ASNase, appearing as a homodimer consisting of 9α helices and 17β strands. Pairwise alignment (Fig. 7) revealed that several amino acid residues from both monomers are responsible for catalysis, such as Thr12,85 (Thr11,85 in T. kodakarensis) and Ser54, Asp86, and Lys156 from the neighboring subunit, as being both essential and highly conserved in microbial l-ASNases [37, 38]. Furthermore, the amino acid Thr12 participates in l-Asn recognition and catalysis between β1 and β2 sheets in a β-hairpin structure (Figs. 5 and 6), which renders it flexible enough to mediate l-ASNase activity [38]. The other key amino acid residue Thr85 localized between α3 and β5 in a coil structure together with Thr12 mediates the sequential nucleophilic reaction during the amidohydrolysis of l-Asn that is involved in the formation of the intermediate compound β-acyl-enzyme, followed by the enzyme-bound water molecule, resulting in the release of aspartic acid and ammonia [38].

Sequence annotation for B. sonorensis l-ASNase showing the location of α-helices and residues contact to ligand and ions. Secondary structure by homology ( ) active sites residues from PDB site record (
) active sites residues from PDB site record ( ); residues contacts to ligand (
); residues contacts to ligand ( ) and to ions (
) and to ions ( ) (Color figure online)
) (Color figure online)

Predicted 3D structure of B. sonorensis l-ASNase monomer structure (a) showing the overall secondary structure and the N- and C-terminus residues (Met1 and Ile329) and homodimer (b) showing important residues involved in catalysis (yellow) (Color figure online)

Pairwise alignment of B. sonorensis l-ASNase and Thermococcus kodakarensis l-ASNase showing residues involved in substrate binding and catalysis (Thr12,85) and highly conserved residues that participate in catalysis and dimer formation (Ser54, Asp86, and Lys156)
3.3 Expression and Purification of Recombinant B. sonorensis l-ASNase
Overnight induction with lactose at a concentration of 2 g/L in the fermentation medium resulted in the formation of soluble active cytosolic protein, as shown in Fig. 9a, Lanes 1 and 2, with a crude enzyme activity of 4588.1 U/mL and a specific activity of 186.58 U/mg protein (Table 3). The fusion recombinant B. sonorensis l-ASNase was purified using single-step nickel affinity resin (Fig. 8b), with a specific activity of 4438.62 U/mg protein, purification of 23.79-fold, and a total yield of 49.37% (Table 3). The purified recombinant l-ASNase yielded a single protein band at 36 kDa on 10% SDS-PAGE (Fig. 8c), which interacted specifically with the anti-His-tag monoclonal antibody, as shown in Fig. 8d.

SDS-PAGE (10%) for recombinant B. sonorenesis l-ASNase expressed in E. coli uninduced culture Panel a (Lanes 3 and 3) and lactose-induced culture (Lanes 1 and 2). b Purification profile of recombinant B. sonorenesis l-ASNase on nickel affinity column. c SDS-PAGE (10%) for nickel affinity–purified recombinant B. sonorenesis l-ASNase (Lanes 2–7). d Western blotting analysis of B. sonorenesis recombinant l-ASNase with anti-His-tag monoclonal antibody (Lanes 9–14). Lanes 5, 1, and 8 represent GangNam-STAIN prestained protein ladder
3.4 Characterization of the Recombinant Enzyme
3.4.1 Optimum Temperature, pH, and Thermostability
The purified l-ASNase exhibited activity at different incubation temperatures from 37 to 60 °C, with the optimum temperature being 45 °C (Fig. 9a). The maximum activity of the purified recombinant l-ASNase was found at pH 7.0 (Fig. 9b). Considering the thermal stability of the purified l-ASNase, the enzyme exhibited temperature stability, ranging from 25 to 45 °C, after an incubation period of 60 min, and the residual activity was found to be 98.84%, 95.0%, and 85.084%, respectively (Fig. 9c). On the other hand, the enzyme retained 22.8% of its original activity after 60-min incubation period at 50 °C.

Optimum temperature (a), pH (b), and thermostability (c) of the purified B. sonorensis l-ASNase. d Effect of the free amino acid glycine on the activity of recombinant B. sonorensis l-ASNase. Buffer systems for pH optimum were as follow: 100 mM potassium acetate buffer for pH 4 and 5, 100 mM potassium phosphate buffer for pH 6 and 7, and 100 mM Tris–HCl buffer for pH 8, 9, and 10
3.4.2 Effect of Metal Ions, Reducing Agents, EDTA, and the Free Amino Acid Glycine
The effect of metal ions (chloride and sulfate forms), reducing agents (DTT and β-mercaptoethanol), metal chelating agent, EDTA, and glycine was also investigated in this study (Table 4 and Fig. 9d). The metal ions in chloride form, namely KCl, NaCl, CaCl2, and MgCl2, increased the enzyme activity at a concentration of 5 mM, whereas the sulfated forms (Na2SO4, ZnSO4, and MgSO4) at the same concentration slightly enhanced the l-ASNase activity (Table 4). Reducing agents, DTT and β-mercaptoethanol, at the respective concentrations of 1 and 5 mM inhibited the activity of l-ASNase. The metal chelating agent EDTA at concentrations of 1 and 5 mM inhibited the activity of purified l-ASNase by 44.92% and 95.34%, respectively. Regarding free amino acids, studies have demonstrated that l-Gly promoted the activity of some l-ASNases such as human l-ASNase-3 (hl-ASNase3) by the cleavage reaction of hl-ASNase3 to α and β peptides [39,40,41]. We investigated the effect of the free amino acid l-Gly on the activity of B. sonorensis l-ASNase and observed that glycine at 500 mM concentration significantly increased the activity of l-ASNase at 4 °C and 25 °C by 31% and 14%, respectively (Fig. 9d).
3.4.3 Determination of Km and Vmax
One of the major and important criteria for l-ASNases to be used as therapeutic drug candidates is an appropriate lower value of the Michaelis constant Km toward l-Asn. The lower the Km value, the better the affinity of l-ASNase for l-Asn and the more efficient the enzyme in the treatment of ALL [10]. The Km and Vmax values were calculated using the reaction substrate l-Asn, and it was found that the recombinant enzyme had Km and Vmax values of 2.004 mM and 3723 µmol min−1, respectively.
The concentration of l-Asn in the serum was within the range of 70–50 µM [42], and because the calculated Km value of recombinant B. sonorensis l-ASNase was comparable to those of E. coli, E. carotovora, and E. chrysanthemi [43, 44], this newly isolated recombinant l-ASNase is a possible potential drug candidate for treating childhood ALL.
3.4.4 Specificity of Recombinant l-ASNase toward Reaction Substrate
The substrate specificity of B. sonorensis recombinant l-ASNase was analyzed by examining different reaction substrates. Results showed that the purified recombinant l-ASNase exhibited remarkable specificity toward the reaction substrate l-Asn; moreover, it exhibited lower activity toward the reaction substrate l-Gln (15.72%), whereas no specificity was observed with urea and acrylamide under identical assay conditions (Table 5). Our results were similar to those of the two major currently marketed l-ASNAases Oncaspar® and Erwinase®, as both have low Km values toward the reaction substrate l-Asn and are also capable of deaminating l-Gln to l-Glu and ammonia.
In conclusion, microbial l-ASNases are being used as protein biopharmaceutical drugs for the treatment of childhood ALL and other blood malignant diseases for several decades. However, the toxicities associated with this drug protein treatment require effective management, continuous requirement of new sources of the enzyme by searching for novel platforms, and development of the existing products to be safer with fewer adverse effects and long lasting in the blood. Overall, the present study has demonstrated for the first time the isolation, characterization, cloning, and overexpression of B. sonorensis l-ASNase; however, the level of the purified recombinant enzyme that can be used as a novel therapeutic strategy for ALL is still under investigation.
References
Saeed H, Soudan H, El-Skarkawy A, Farag A, Embaby A, Ataya F (2018) Expression and functional characterization of Pseudomonas aeruginosa recombinant L. asparaginase. Protein J 37:461–471
El-Naggar N, Moawad H, El-Shweihy NM, El-Ewasy SM, Elsehemy IA, Abdelwahed NAM (2020) Process development for scale-up production of a therapeutic L-asparaginase by Streptomyces brollosae NEAE-115 from shake flasks to bioreactor. Sci Rep 9:1–20
Cachumba JJM, Antunes FAF, Peres GFD, Brumano LP, Santos JCD, Da Silva SS (2016) Current applications and different approaches for microbial L-asparaginase production. Braz J Microbiol 47:77–85
Mohideen AKS (2020) Molecular docking study of L-Asparaginase I from Vibrio campbellii in the treatment of acute lymphoblastic leukemia (ALL). The EuroBiotech J 4:8–16
Asselin B, Rizzari C (2015) Asparaginase pharmacokinetics and implications of therapeutic drug monitoring. Leuk Lymphoma 56:2273–2280
John C, Herz T, Boos J, Langer K, Hempel G (2016) Asymmetrical flow field-flow fractionation for the analysis of PEG-asparaginase. Talanta 146:335–339
Becker FF, Broome JD (1969) L-Asparaginase: inhibition of endogenous RNA polymerase activity regenerating liver. Arch Biochem Biophys 130:332–336
Nguyen HA, Su Y, Zhang JY, Antanasijevic A, Caffrey M, Schalk AM, Liu L, Rondelli D, Oh A, Mahmud DL (2018) A novel L-Asparaginase with low L-Glutaminase coactivity is highly efficacious against both T-and B-cell acute lymphoblastic leukemias in vivo. Cancer Res 78:1549–1560
Panosyan EH, Grigoryan RS, Avramis IA, Seibel NL, Gaynon PS, Siegel SE, Fingert HJ, Avramis VI (2004) Deamination of glutamine is a prerequisite for optimal asparagine deamination by L-Asparaginases in vivo (CCG-1961). Anticancer Res 24:1121–1126
Ollenschläger G, Roth E, Linkesch W, Jansen S, Simmel A, Mödder B (1988) L-Asparaginase-induced derangements of glutamine metabolism: the pathogenetic basis for some drug-related side-effects. Eur J Clin Invest 18:512–516
Hawkins DS, Park JR, Thomson BG, Felgenhauer JL, Holcenberg JS, Panosyan EH, Avramis VI (2004) L-Asparaginase pharmacokinetics after intensive polyethylene glycol-conjugated L-Asparaginase therapy for children with relapsed acute lymphoblastic leukemia. Clin Cancer Res 10:5335–5341
Chan WK, Lorenzi PL, Anishkin A, Purwaha P, Rogers DM, Sukharev S, Rempe SB, Weinstein JN (2014) The glutaminase activity of L-Asparaginase is not required for anticancer activity against ASNS-negative cells. Blood 123:3596–3606
Parmentier JH, Maggi M, Tarasco E, Scotti C, Avramis VI, Mittelman SD (2015) Glutaminase activity determines cytotoxicity of L-Asparaginases on most leukemia cell lines. Leuk Res 39:757–762
Offman MN, Krol M, Patel N, Krishnan S, Liu J, Saha V, Bates PA (2011) Rational engineering of L-Asparaginase reveals importance of dual activity for cancer cell toxicity. Blood 117:1614–1621
Kumar K, Kaur J, Walia S, Pathak T, Aggarwal D (2014) L-asparaginase: an effective agent in the treatment of acute lymphoblastic leukemia. Leuk Lymphoma 55:256–262
Hijiya N, Van Der Sluis IM (2016) Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk Lymphoma 57:748–757
Kloos RQH, Uyl-de Groot CA, van Litsenburg RRL, Kaspers GJL, Pieters R, van der Sluis IM (2017) A cost analysis of individualized asparaginase treatment in pediatric acute lymphoblastic leukemia. Pediatr Blood Cancer 64:e26651
Heitink-Pollé KM, Prinsen BH, de Koning TJ, van Hasselt PM, Bierings MP (2013) High incidence of symptomatic hyperammonemia in children with acute lymphoblastic leukemia receiving pegylated L-Asparaginase. JIMD Rep 7:103–108
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Imada A, Igarasi S, Nakahama K, Isono M (1973) Asparaginase and glutaminase activities of micro-organisms. Microbiol 76:85–99
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76:4350–4354
Saeed H, Ali H, Soudan H, Embaby A, El-Sharkawy A, Farag A, Hussein A, Ataya F (2018) Molecular cloning, structural modeling and production of recombinant Aspergillus terreus L. asparaginase in Escherichia coli. Int J Biol Macromol 106:1041–1051
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M (2008) Phylogeny. fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469
Milburn D, Laskowski RA, Thornton JM (1998) Sequences annotated by structure: a tool to facilitate the use of structural information in sequence analysis. Protein Eng 11:855–859
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303
Guex N, Peitsch MC, Schwede T (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30:S162–S173
Saeed H, Hemida A, El-Nikhely N, Abdel-Fattah M, Shalaby M, Hussein A, Eldoksh A, Ataya A, Aly A, Labrou N, Nematalla H (2020) Highly efficient Pyrococcus furiosus recombinant L-Asparaginase with no glutaminase activity: expression, purification, functional characterization, and cytotoxicity on THP-1, A549 and Caco 2 cell lines. Int J Biol Macromol 156:812–828
Chohan SM, Rachid N (2013) TK1656, a thermostable L-asparaginase from Thermococcus kodakaraensis, exhibiting highest ever reported enzyme activity. J Biosci Bioeng 116:438–443
Emadi A, Law JY, Strovel ET, Lapidus RG, Jeng LJB, Lee M, Blizer MG, Carter-Cooper BA, Sewell D, Van Der Merwe I, Philip S, Imran M, Yu SL, Li H, Amrein PC, Duong VH, Sausville EA, Baer MR, Fathi AT, Singh Z, Bentzen SM (2018) Asparaginase Erwinia chrysenthemi effectively depletes plasma glutamine in adult patients with relapsed refractory acute myeloid leukemia. Cancer Chemother Pharmacol 81:217–222
Derst C, Henseling J, Röhm KH (2000) Engineering the substrate specificity of Escherichia coli asparaginase II. Selective reduction of glutaminase activity by amino acid replacements at position 248. Protein Sci 9:2009–2017
Stark RM, Suleiman MS, Hassan IJ, Greenman J, Millar MR (1997) Amino acid utilization and deamination of glutamine and asparagine by Helicobacter pylori. J Med Microbiol 46:793–800
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+:archtecture and applications. BMC Bioinform 10:421–430
Remmert M, Biegert A, Hauser A, Söding J (2012) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 9:173–175
Guo J, Coker AR, Wood SP, Cooper JB, Chokan SM, Rashid N, Akhtar M (2017) Structure and function of the thermostable L- asparaginase from Thermococcus kodakarensis. Acta Crystallogr D Struct Biol 73:889–895
Verma N, Kumar K, Kaur G, Anand S (2007) L-asparaginase: a promising chemotherapeutic agent. Crit Rev Biotechnol 27:45–62
Nomme J, Su Y, Lavie A (2014) Elucidation of the specific function of the conserved threonine triad responsible for human L-Asparaginase autocleavage and substrate hydrolysis. J Mol Biol 426:2471–2485
Su Y, Karamitros CS, Nomme J, McSorley T, Konrad M, Lavie A (2013) Free glycine accelerates the autoproteolytic activation of human L-Asparaginase. Chem Biol 20:533–540
Tikkanen R, Riikonen A, Oinonen C, Rouvinen R, Peltonen L (1996) Functional analyses of active site residues of human lysosomal aspartylglucosaminidase: implications for catalytic mechanism and autocatalytic activation. EMBO J 15:2954–2960
Cooney D, Capizzi R, Handschumacher R (1970) Evaluation of L-Asparagine metabolism in animals and man. Cancer Res 30:929–935
Kotzia GA, Labrou NE (2007) L-Asparaginase from Erwinia chrysanthemi 3937: cloning, expression and characterization. J Biotechnol 127:657–669
Ho PPK, Milikin EB, Bobbitt JL, Grinnan EL, Burk PJ, Frank BH, Beck LD, Squires SW (1970) Crystalline L-asparaginase from Escherichia coli B. I. Purification and chemical characterization. J Biol Chem 245:3708–3715
Funding
This research has been funded by Academy of Scientific Research and Technology (ASRT), Grant No. 35d/2018.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aly, N., El-Ahwany, A., Ataya, F.S. et al. Bacillus sonorensis L. Asparaginase: Cloning, Expression in E. coli and Characterization. Protein J 39, 717–729 (2020). https://doi.org/10.1007/s10930-020-09932-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-020-09932-x