
Abstract
The present investigation was conducted to evaluate salt tolerance in ten genotypes of soybean (Glycine max L.). Twelve-day-old seedlings, grown hydroponically, were treated with 0, 25, 50, 75, 100, 125 and 150 mM NaCl for 10 days. Growth, lipid peroxidation and antioxidant enzyme activities were evaluated. Growth, measured in terms of length, fresh weight and dry weight of plants, was drastically reduced in Pusa-24 while there was little effect of NaCl treatment on Pusa-37 genotype of soybean. High level of lipid peroxidation was observed in Pusa-24 as indicated by increased level of malondialdehyde. Activities of superoxide dismutase, catalase, ascorbate peroxidase and glutathione reductase were maximum in Pusa-37 where 9-, 1-, 5- and 6-fold increase over control were observed, respectively. The results suggested that Pusa-24 and Pusa-37 are salt-sensitive and salt-tolerant genotype of soybean, respectively, and antioxidant defence system is involved in conferring the sensitiveness and tolerance in these genotypes. Salt-tolerant genotype Pusa-37, was further analysed by 2-dimensional gel electrophoresis to analyse the differential expression of proteins at high salt stress. In the present study, 173 protein spots were identified. Of these, 40 proteins were responsive to salinity in that they were either up- or downregulated. This study could help us in identifying the possible regulatory switches (gene/s) controlling novel proteins of the salt-tolerant genotype of the crop plants and their possible role in defence mechanism.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Over 800 million ha of land throughout the world are salt affected, either by salinity (397 million ha) or the associated condition of sodicity (434 million ha) [1]. This is over 6 % of the world’s total land area. However, a significant proportion of recently cultivated agricultural land has become saline because of land clearing or irrigation. Of the 1,500 million ha of land farmed by dry land agriculture, 32 million (2 %) are affected by secondary salinity to varying degrees. Of the current 230 million ha of irrigated land, 45 million ha are salt affected (20 %) [1]. Soil salinity continues to be one of the world’s most serious environmental problems in agriculture. It limits agricultural production throughout the world. Salt stress induces various biochemical and physiological responses in plants and affects almost all plant processes [2]. Salt stress can affect plant survival, biomass, plant height and plant form, where such changes in morphology affect the capacity of a plant to collect light, water and nutrients [3].
Genetic variability within a species offers a valuable tool for studying mechanisms of salt tolerance. Soybean (Glycine max (L.) Merr.) is the first largest oil seed crop in India and played a major role in upliftment of socio-economic status of Indian farmers. Indian soybean has to be globally competitive in the present era of opening up world trade and liberalised economy. India produced 5.60 mt of soybean in an area of 6.0 mha during 2001–2002 with an average of 0.93 t/ha and contributed 3.17 % of total global production [4]. Soybean-producing regions in India range from the lower Himalayan Hills and Northern Plain in the north to the Deccan plateau. The soybean varieties cultivated in these areas were developed through separate breeding programmes. Identification of diverse genotypes is the prerequisite for improvement of any trait in the crop plants. Over the past few years, much attention has been concentrated on resolving the identity of salt-stress proteins, in order to identify and understand the role of proteins in soybean salt tolerance. However, at present much of their functions are still unclear. Studying differential responses of genotypes in terms of antioxidant defence system and proteins alteration, with contrasting stress tolerance will help to dissect the underlying salt-stress tolerance mechanisms. In the present study, a concerted effort was made firstly to study the physiological response of soybean genotypes under salt stress by analysing antioxidative defence mechanism and secondly, to analyse differential protein expression of salt-tolerant genotype by using 2-dimensional gel electrophoresis.
Materials and Methods
Seeds of soybean genotypes (Pusa-20, Pusa-40, Pusa-37, Pusa-16, Pusa-24, Pusa-22, BRAGG, PK-416, PK-1042 and DS-9712) were obtained from the Indian Agricultural Research Institute, New Delhi, India. These genotypes were surface sterilised and sown in soilrite (Soilrite Mix, Keltech Energies Limited, Bangalore). After 3 days of proper germination, the seedlings were transferred to Hoagland’s solution [5] with the following composition of macro- and micronutrients (in mM), 2.4 Ca (NO3)2, 1.0 KH2PO4, 3.0 KNO3, 1.0 MgSO4 and 0.5 NaCl and (in μM) 23.1 H3BO3, 4.6 MnCl2, 0.38 ZnSO4, 0.16 CuSO4, 0.052 H2MoO4 and 44.8 FeSO4 (as ferric sodium-ethylenediaminetetraacetic (EDTA) complex) in hydroponic culture system. The growth chamber was maintained at a photosynthetic photon flux density of 430 μmol m−2 s−1, 14 h of light, 10 h of dark and a relative humidity of 60 %. The nutrient solution was bubbled with sterile air and changed on alternate days. Ten-day-old seedlings were given seven levels of salt in the form of NaCl viz., T1 = 0 mM NaCl, T2 = 25 mM NaCl, T3 = 50 mM NaCl, T4 = 75 mM NaCl, T5 = 100 mM NaCl, T6 = 125 mM NaCl and T7 = 150 mM NaCl. Growth performance, anti-oxidant defence mechanism and proteome expression were studied after 12 days of salt treatment.
Determination of Fresh Weight, Dry Weight and Length of Plant
Fresh weights (FW) of shoots and roots were weighed on an electronic top pan balance (Model BL-210-S, Sartorius, Germany). For dry weight (DW), determination samples were oven dried at 65 ± 2 °C for 72 h and then weighed independently. FW and DW were expressed in grammes per plant. Length of the plant was measured by using a metric scale and expressed in centimetre.
Lipid Peroxidation
The level of lipid peroxidation in the leaves was determined as malondialdehyde (MDA) content by the method of Heath and Packer [6]. Fresh tissue was ground in 0.1 % trichloroacetic acid (TCA) with a mortar and pestle and centrifuged at 10,000 rpm for 5 min; 1.0 ml of supernatant was taken in a separate test tube, to which 4.0 ml of 0.5 % thiobarbituric acid (TBA; w/v) was added. The mixture was heated at 95 °C for 30 min. It was then quickly cooled in an ice bath and re-centrifuged at 5,000 rpm for 5 min to suspend the turbidity. The absorbance of the supernatant was read at 532 and 600 nm, corrected for unspecific turbidity by subtracting the value at 600 nm. TBA reagent was used as blank. The concentration of MDA was calculated using an extinction (ε) of 155 mM−1 cm−1 and expressed as nanomoles per gramme of FW.
Estimation of Proline Content
To determine free proline level, 0.5 g of leaf samples from each group were homogenised in 3 % (w/v) sulphosalycilic acid and then homogenate filtered through filter paper [7]. Mixture was heated at 100 °C for 1 h in water bath after addition of 2 ml of 1 % ninhydrin and 2 ml of 75 % glacial acetic acid. Reaction was then stopped by ice bath. The mixture was extracted with toluene and the absorbance of fraction with toluene aspired from liquid phase was read at 520 nm. Proline concentration was determined using calibration curve and expressed as micromoles proline per gramme of FW.
Enzyme Assays
Leaves were ground with a mortar and pestle in liquid nitrogen and homogenised with ice-cold 50-mM sodium phosphate buffer (pH 6.8) containing 1 mM EDTA and 2 % (w/v) polyvinylpolypyrrolidone. The whole extraction procedure was carried out at 4 °C. The homogenate was centrifuged at 13,000×g for 40 min at 4 °C and supernatant used for assays of the activities of superoxide dismutase (SOD), ascorbate peroxidase (APX), catalase (CAT) and glutathione reductase (GR). In vitro assay of SOD activity was determined by monitoring its ability to inhibit photochemical reduction of nitroblue tetrazolium (NBT) at 560 nm [8]. The assay mixture consists of 1 ml of reaction buffer, 1 M sodium bicarbonate, 200 mM methionine, 3 mM EDTA, 60 μM riboflavin and 100 μl of enzyme extract in a test tube. Test tubes were shaken and placed 30 cm from light bank consisting of six 15 W fluorescent lamps. The reaction was allowed to run for 10 min and stopped by switching the light off. The reduction in NBT was followed by reading absorbance at 560 nm. Blanks and controls were run the same way but without illumination and enzyme, respectively. One unit of SOD was defined as amount of enzyme which produced a 50 % inhibition of NBT reduction under the assay conditions [9]. The activity was expressed in enzyme unit (EU) per milligramme protein per hour.
In vitro assay of APX activity was estimated by the method used by Nakano and Asada [10]. To assay enzyme activity, the rate of H2O2-dependent oxidation of ascorbic acid was determined. Reaction mixture contained 1.5 ml of the reaction buffer, 0.1 ml of 0.3 % H2O2 (v/v), 0.1 ml of the reaction buffer, 0.1 ml of 0.3 % H2O2, 0.1 ml of 0.5 mM ascorbate, 3 mM of 0.1 m of EDTA and 100 μl of the enzyme extract. APX activity was determined spectrophotometrically by monitoring the decrease in ascorbate at A290 (ε = 2.8 mM−1 cm−1) as described by Nakano and Asada [10]. One EU determines the amount of enzyme necessary to decompose 1 μmol ascorbate mg−1 of protein min−1 at 25 °C and expressed as EU per milligrammes of protein.
In vitro activity of CAT was determined by the method of Aebi [11]. The activity was determined by monitoring the disappearance of H2O2, measuring a decrease in the absorbance at 240 nm. The reaction was carried in a reaction mixture containing 1.0 ml of the reaction buffer, 0.1 ml of EDTA, 0.1 ml of the enzyme extract, 0.1 ml of H2O2 and allowed to run for 3 min. The enzyme activity was calculated using the extinction coefficient 0.036 mM−1 cm−1. One EU determines the amount of enzyme necessary to decompose 1 μmol of H2O2 mg−1 protein min−1 at 25 °C and expressed as EU milligrammes protein.
In vitro assay of GR activity was determined by the method of Foyer and Halliwell [12]. The GR activity was determined by stoichometric conversion of nicotinamide adenine dinucleotide phosphate (NADPH) at the baseline level of NADPH absorbance at 340 nm. The reaction mixture contained 1 ml reaction buffer (0.2 mM NADPH and 0.5 mM oxidised glutathione (GSSG)) and 0.1 ml of the enzyme extract. The activity was calculated by using extinction coefficient of 6.2 mM−1 cm−1 and expressed as EU per milligramme of protein.
Protein concentration was determined according to Bradford [13], using bovine serum albumin (BSA) as a standard.
Proteome Analysis
Protein Extraction
The 12-day-old seedlings of Pusa-37 (salt tolerant) genotype of soybean were treated with six different NaCl treatments (0, 25, 50, 75, 100, 125 and 150 mM NaCl for 10 days). Proteins were extracted using the method of Damerval et al. [14]. The leaf tissue (5 g) was frozen in liquid N2 and ground to a fine powder using a ceramic mortar and pestle; 1.0 g of the resulting powder was suspended in 5 ml of chilled (4 °C) extraction buffer containing 175 mmol l−1 tris(hydroxymethyl) aminomethane hydrochloride (pH 8.8), 50 gl−1 sodium dodecyl sulphate (SDS), 150 ml l−1 glycerol, 0.70 ml l−1 2-mercaptoethanol and 10 ml l−1 plant protease inhibitor mix, and grinding was maintained for an additional 30 s. Homogenised cell debris was removed by filtering the homogenate through two layers of Miracloth and centrifuged at low speed (500 ×g) at 4 °C for 15 min. The supernatant was collected and mixed by vortexing with 20 ml of cold acetone containing 100 g l−1 TCA and 0.70 ml l−1 2-mercaptoethanol and was kept at −20 °C for at least 1 h to allow protein precipitation. Precipitated proteins were centrifuged at 15,000×g at 4 °C for 45 min, and the pellet obtained was washed three times with a cold water/acetone solution (20:80, v/v) containing 0.70 ml l−1 2-mercaptoethanol and between rinses was centrifuged at 15,000×g for 15 min. Successively, the supernatant was removed and the pellet was slowly dried under nitrogen and resuspended in the isoelectric focusing (IEF) extraction solution consisting of 9 mol l−1 urea, 20 g l−1 3-[(3-cholamidopropyl)dimethylammonio]-1-propane sulphonate, 30 g l−1 dithiothreitol (DTT) and 20 ml l−1 at pH 4–7 ampholytes. In order to obtain a complete protein solubilisation, the sample was incubated for 2 h at 33 °C. Finally, the sample was centrifuged at 15,000×g at 4 °C for 30 min, and the supernatant was subjected to IEF. The protein was quantified using 2D Bradford kit (Bio-Rad Laboratories, Hercules, CA) with BSA as the standard.
Two-Dimensional Electrophoresis
Two-dimensional electrophoresis (2-DE) of proteins was performed in accordance with the method of O’Farrel [15] with some modifications. Briefly, 450 μl of solution containing 500 μg of proteins was applied in the strip holder and immobilised pH gradient strips (7 cm, pH 4–7) were placed and covered with mineral oil. IEF was carried out by using an IEF system (Protean IEF Cell, Bio-Rad), applying the following conditions. For the rehydration step, the voltage was maintained for 12 h at 30 V, then the proteins were focused for 1 h at 500 V, 1 h at 1,000 V and 8 h and 20 min at 8,000 V. The temperature was maintained at 20 °C and the current was 50 μA per strip. After IEF, the strips were equilibrated in DTT solution (Bio-Rad) followed by iodoacetamide solution (Bio-Rad) as described by Chivasa et al. [16] and then stored at −20 °C. The second-dimension separation of proteins was performed according to the method of Laemmli [17] on a 12.5 % SDS polyacrylamide gel using Mini Protean tetra cell electrophoresis unit (Bio-Rad). The electrophoresis was carried out at 25 °C and 2.5 W/gel for 30 min and then 17 W/gel for 5 h and 40 min until the bromophenol blue dye front arrived at the bottom of the gels. Following SDS polyacrylamide gel electrophoresis (PAGE), gels were stained with Coomassie R 250 in accordance with the manufacture’s manual (Bio-Rad). Molecular weights of the proteins were compared with reference to broad-range protein marker (Bio-Rad) of known molecular weights used.
Gel Image and Data Analysis
Two-dimensional (2D) wet gels were scanned by a Bio-Rad GS 710 Calibrated Imaging Densitometer and the comparison of protein patterns was done automatically using the ImageMaster™ PDQuest software (version 8.0) of Bio-Rad. The optimised parameters were as follows: saliency, 2.0; partial threshold, 4; and minimum area, 50. The intensity of the spots was normalised to that of landmark proteins used for internal standardisation. Spots were quantified on the basis of their relative volume, which was determined by the ratio of the volume of a single spot to the whole set of spots.
Scoring Methods and Statistical Analyses
Spots were detected and quantified by the Gaussian method. The groups were defined after aligning and matching. PDQuest automatically computes the quantification values in per cent of volume (%volume). For each matched spot, the %volume was calculated as its volume divided by the total volume of matched spots (referred to hereafter by intensity). To compare the intensity of each polypeptide, the following model was run using the PROC GLM of SAS Statistical Package (1990),
where V i is the variety effect, N j is the N level effect, VN ij is the interaction between variety and N level, B k is the repetition or block effect, NB jk is the interaction between treatment and block and E ijk is the residual effect. All the effects were tested against the residual (E ijk) except the treatment effect, which was tested against NB jk in order to take into account the split-plot structure.
Only those with significant (quantitative changes more than 2-fold in abundance) and reproducible changes in three replicates were used for further analysis.
Mass Spectroscopy Analysis
-
(a)
In-gel digestion
The excised gel spots were destained by 100 μl of destain solution (30 mM potassium ferricyanide (Sigma) in 100 mM sodium thiosulfate (Merck)) with shaking for 5 min. After the solution was removed, the gel spots were incubated with 200 mM ammonium bicarbonate (Sigma) for 20 min. The gel pieces were dried in a speed vacuum concentrator for 5 min and then rehydrated with 20 μl of 50 mM ammonium bicarbonate containing 0.2-μg modified trypsin (Sigma) for 45 min on ice. After removal of solution, 30 μl of 50-mM ammonium bicarbonate was added and the digestion was performed overnight at 37 °C.
-
(b)
LC-MS/MS analysis
For analyses by MS/MS, 15 μl of the peptide solutions from the digestion supernatant was diluted with 30 μl in 5 % formic acid, loaded onto the column and washed with 30 μl of 5 % formic acid. Peptides were eluted with 2.0 μl methanol/water/formic acid (50/49/1, v/v/v) directly into a pre-coated borosilicate nanoelectrospray needles (EconoTipTM, New Objective). MS/MS of peptides generated by in-gel digestion was performed by nano-ESI on a Q-TOF2 mass spectrometer (Micromass, Manchester, UK). The source temperature was 80 °C. A potential of 1 kV was applied to the pre-coated boron silicate nano-ESI needles in the ion source combined with a nitrogen back-pressure of 0–5 psi to produce a stable flow rate (10–30 nl/min). The mass spectrometer operated in an automatic data dependent MS/MS to collect ion signals from the eluted peptides. In this mode, the most abundant peptide ion peak with doubly or triply charged ion in a full-scan mass spectrum (m/z 400–1,500) was selected as the precursor ion. Finally, an MS/MS spectrum was recorded to confirm the sequence of the precursor ion using collision-induced dissociation with a relative collision energy dependant on molecular weight. The cone voltage was 40 V. The quadrupole analyser was used to select precursor ions for fragmentation in the hexapole collision cell. The collision gas was Ar at a pressure of 6–7 × 10–5 mbar, and the collision energy was 20–30 V. Product ions were analysed using an orthogonal TOF analyser, fitted with a reflector, a microchannel plate detector and a time-to-digital converter. The data were processed using a Mass Lynx Windows NT PC system. To identify the protein, peptide masses from matrix-assisted laser desorption ionisation time-of-flight mass spectrometry (MALDI-TOF-MS) were matched with the theoretical molecular weight of peptides for proteins in the NCBI database using MASCOT software. Also, all MS/MS spectra recorded on tryptic peptides derived from spot were searched against protein sequences from NCBInr and EST databases using the MASCOT search program (www.matrixscience.com).
Statistical Analysis
All analyses were done on a completely randomised design. All data obtained were subjected to one-way analyses of variance, and the mean differences were compared by lowest standard deviations test. Each data point was the mean of six replicates (n = 6) and comparisons with p values of <0.05 were considered significantly different (Table 1). In all the figures, the spread of values is shown as error bars representing standard errors (SE) of the means
Results
Growth Parameters
Growth of soybean genotypes was measured in terms of plant shoot length and root length, plant FW and DW on day 12 of salt treatment. Enormous variability in the growth of soybean genotypes was observed under various levels of salt treatments. The growth of Pusa-24, PK-416 and Pusa-20 was significantly reduced at all the NaCl treatments. The reduction in the plant length (shoot and root), FW and DW of Pusa-24 was 23–67, 32–73, 22–36 and 20–85 %. PK-416 was 19–41, 12–31, 21–80 and 46–95 %, respectively, and that of Pusa-20 was 12–20, 11–19, 5–68 and 24–77 %, respectively. There was no significant effect of any NaCl treatment on the growth of Pusa-37. The growth of BRAGG and PK-1042 was only affected significantly at 150 mM NaCl treatment (T6). Pusa-16, Pusa-22, Pusa-40 and DS-9712 were able to tolerate NaCl treatment up to the level of 75 mM (T3) as observed in terms of growth (Tables 1 and 2).
Proline Accumulation
Proline accumulation in the soybean genotypes increased significantly with increasing levels of NaCl treatments, except Pusa-24. Genotype PK-416 and Pusa-20 showed significant increase only at T1 (25 mM NaCl). The percent increase in the proline accumulation was 94–188 in Pusa-37 followed by BRAGG 64–178 and PK-1042 26–79 %, respectively by NaCl treatments when compared with control. Maximum increase was observed at T6 (150 mM NaCl) treatment (Fig. 1).

Proline accumulation (in nanomoles per gramme of FW) in the soybean genotypes at different NaCl concentrations
Lipid Peroxidation
In order to evaluate the level of lipid peroxidation caused by oxidative stress generated by saline conditions, MDA level was determined in soybean genotypes subjected to different level of NaCl for 12 days. Maximum amount of the MDA (2.5-fold) was reported in Pusa-24 followed by PK-416 and Pusa-20, which is significant at all levels of NaCl treatments, compared with control. The MDA level in Pusa-37, BRAGG and PK-1042 were 10, 20 and 25 %, respectively higher over control. The percent increase of MDA in Pusa-16, Pusa-22, Pusa-40 and DS-9712 was 62, 75, 61 and 57 %, respectively when compared with control (Fig. 2).

MDA Content (in nanomoles per gramme of FW) in the soybean genotypes at different NaCl concentrations
Antioxidant Enzymes
Activity of SOD showed large variation in soybean genotypes at various levels of NaCl treatments. There was up to 9-fold increase in the activity of SOD in Pusa-37, BRAGG and PK-1042 by NaCl treatments over control. No significant effect of NaCl treatment was reported in SOD activity of Pusa-24, PK-416 and Pusa-20, when compared with control. The SOD activity increased significantly in Pusa-16 (29 %), Pusa-22 (21 %), Pusa-40 (22 %) and DS-9712 (19 %) with all level of NaCl treatment when compared with control. However, maximum SOD activity was reported at T3 (75 mM NaCl) (Fig. 3).

SOD activity (EU; in milligrammes of protein) in the soybean genotypes at different NaCl concentrations
Salt (NaCl) treatment induced the CAT activity differently in soybean genotypes. Maximum induction (18–337 %) was observed in Pusa-37 by NaCl treatments. There was no significant effect of NaCl treatments on the CAT activity in Pusa-24, PK-416 and Pusa-20, when compared with control. The activity of CAT was increased by 19–36, 21–35, 18–79 and 19–55 % in Pusa-16, Pusa-22, Pusa-40 and DS-9712, respectively with the increase in the level of NaCl up to 75 mM (T3). The highest CAT activity was observed under (100–150 mM) NaCl stress in these genotypes (Fig. 4).

CAT activity (EU; in milligrammes of protein) in the soybean genotypes at different NaCl concentrations
APX activity showed large variation in soybean genotypes following NaCl treatments. Maximum APX activity was induced in Pusa-37 (3-fold) followed by BRAGG (1.7-fold) and PK-1042 (1.3-fold) with the increasing levels of NaCl treatments. PK-416 and Pusa-20 did not show any significant change in APX activity by NaCl treatments when compared with control. The APX activity increased significantly in Pusa-16 (19 %), Pusa-22 (15 %), Pusa-40 (14 %) and DS-9712 (17 %) with all levels of NaCl treatments. The maximum activity was observed with T3 treatment (Fig. 5).

APX activity (EU; in milligrammes of protein) in the soybean genotypes at different NaCl concentrations
There was a significant increase in GR activity by NaCl treatments in all genotypes except PK-416 and Pusa-20. Maximum increase in GR activity was observed in Pusa-37 (6-fold), followed by BRAGG (5-fold) and PK-1042 (3.1-fold) by NaCl treatments. The GR activity in Pusa-16, Pusa-22, Pusa-40 and DS-9712 was in the range of 22–58, 26–69, 26–86, 23–58 %, respectively by NaCl treatments, when compared with control (Fig. 6).

GR activity (EU) (mg-1protein) in the soybean genotypes at different NaCl concentrations
Proteome Analysis
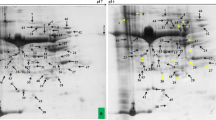
To investigate the response of Pusa-37 (salt tolerant) genotype of soybean to salt stress, 12-day-old untreated and 150-mM NaCl-treated seedlings were used for proteome analysis. Proteins were extracted from the leaves of the seedlings, separated by 2D PAGE and stained with Coomassie brilliant blue. Digital image analysis revealed 173 protein spots (Fig. 7). Of these, 40 proteins were responsive to salinity in that they were either up- or downregulated. Nineteen protein spots (3, 4, 7, 14, 15, 17, 19, 25, 27, 28, 31, 32, 33, 34, 35, 36, 37, 39 and 40) consistently showed significant and reproducible changes in their levels (2- to 7-fold), six protein spots (2, 8, 11, 10, 22 and 23) were downregulated and 14 protein spots (1, 5, 6, 9, 12, 13, 16, 18, 21, 24, 26, 29, 30 and 38) showed no significant changes in their expression level under high salt treatment in each replicate experiment (Fig. 8).

Representative 2-DE maps of soybean leaf proteins; 20 days old soybean seedlings were treated with 150 mM NaCl for 24 h. Total leaf proteins were extracted and separated by 2-DE. In IEF, 150 μg of proteins were loaded onto pH 4–7 IPG strips (7 cm, linear). SDS-PAGE was performed with 12.5 % gels. The spots were visualised by Comassie brilliant blue staining. Differentially expressed protein spots are indicated by arrows in map of control sample (a). Nineteen upregulated spots are indicated in the map by upward arrows in NaCl-treated sample (b), six down regulated spots are indicated on the map by downward arrow NaCl-treated sample (b). The protein spots that are not affected significantly are encircled in NaCl-treated sample (b)

Expression changes of some soybean leaf proteins after exposure to the 150 mM NaCl stress. Histograms represented leaf spot volumes of control and treated samples. Data were presented as means of relative spot volumes ± SE
Identification of Differentially Expressed Proteins
Twenty protein spots with significant changes in their expression profile were excised manually from 2D gels for tryptic digest and subjected to mass spectrometry. To identify the protein, peptide masses from MALDI-TOF-MS were matched with the theoretical molecular weight of peptides for proteins in the NCBI database using MASCOT software (www.matrixscience.com) (Table 3).
Discussion
Soil salinity is the major factor limiting crop yield and productivity in the world over. Also in India intensive agronomic practices, poor water management, irrigation without sufficient drainage systems, long periods of hot and dry seasons and high levels of evaporation lead to the salinisation of almost 2.8 million ha of agricultural land [18]. Shoot and root growth inhibition is a common response to salinity, and plant growth is one of the most important agricultural indices of salt-stress tolerance as indicated by different studies [19, 20]. Our results showed that there was large variability in the response of soybean genotypes to salt treatments in terms of their length, DW and FW. The genotypes, PK-416 and Pusa-20 appeared to be the most salt-sensitive genotypes as inferred from their significantly reduced length, FW and DW in response to the exposure to NaCl. Increased in reductions of these parameters were observed with increased levels of NaCl treatments. Pusa-37 appeared to be the most tolerant soybean genotype since no significant effect of NaCl treatment on growth was found. The growth of BRAGG and PK-1042 was reduced significantly only by the 150 mM NaCl treatment (T6). Pusa-16, Pusa-22, Pusa-40 and DS-9712 showed their tolerance behaviour of up to 75 mM NaCl. Biomass differences among plant species under saline conditions are important in determining tolerance. Similar to our findings, growth was less affected in salt-tolerant sugar beet and moderately salt-tolerant cotton [21]. To work out the physiological mechanism of this variability in soybean genotypes in response to various levels of salt treatments, we have investigated the level of lipid peroxidation and possibility of the role antioxidant defence system in conferring the salt sensitiveness and salt tolerance in these genotypes. Lipid peroxidation, measured as the amount of MDA, is produced when polyunsaturated fatty acids in the membrane undergo oxidation by the accumulation of free oxygen radicals. As lipid peroxidation is the symptom mostly ascribed to oxidative damage, it is often used as an indicator of increased damage [22, 23]. The results reported here showed that the level of MDA was maximum in PK-416 and Pusa-20, moderate in Pusa-16, Pusa-22, Pusa-40 and DS-9712 and minimum in Pusa-37, BRAGG and PK-1042. There are reports of higher increase in the amount of MDA with the increase in salt stress in the salt-sensitive cultivar as compared with tolerant cultivar of rice and in roots of Lemna minor [24–28]. Reactive oxygen species (ROS) disrupt permeability of plasma membrane through peroxidation of lipid in plant tissues [22–24, 26, 29]. There are evidences showing that membrane injury under salt stress is related to increased production of highly toxic ROS [22, 23, 30]. Growth inhibition under salt stress in the most-sensitive soybean genotypes is in good correlation with increased peroxidation levels. Low MDA content was remarkable in the most-tolerant soybean genotypes, Pusa-37. Parallel to our results, low MDA content is important in terms of salt tolerance as represented in different studies. Sairam et al. [31]
There are reports showing that the variability in level of lipid peroxidation is due to the difference in the efficiency of plants to scavenge ROS through efficient antioxidant defence system. Sairam et al. [31]. Here, we have reported that the antioxidant defence system was induced by salinity stress in soybean genotypes; however, there was distinct variability in the induction of antioxidant enzymes. Superoxide ion is scavenged by SOD to produce H2O2, which is subsequently eliminated mainly by APX and CAT. SOD catalyses the conversion of the superoxide anion (O2 −) to hydrogen peroxide (H2O2), which is very damaging to the chloroplasts, nucleic acids and proteins. All the NaCl treatments in our study increased the activity of SOD. There was an up to 9-fold enhancement in the SOD activity of salt-tolerant soybean genotype (Pusa-37). While there was no significant change in the SOD activity of salt-sensitive genotypes (PK-416 and Pusa-20) during NaCl stress, the SOD activity in Pusa-16, Pusa-22, Pusa-40 and DS-9712 increased with increasing levels of NaCl up to 75 mM (T3) when compared with control. These results showed that these genotypes are moderately tolerant to NaCl stress. Earlier studies suggested that the increased SOD activity enables the plant to resist the potential oxidative damage caused by NaCl salinity exposure [23, 25, 27].
CAT is the main scavenger of H2O2 in peroxisomes, converting it to water and molecular oxygen [32]. Under salt stress, a significant dose-dependent increase in CAT activity was observed in salt-tolerant Pusa-37. Interestingly, CAT activity was not affected significant by any levels of NaCl treatments in BRAGG, PK-1042, PK-416 and Pusa-20 soybean genotypes. CAT activity of Pusa-16, Pusa-22, Pusa-40 and DS-9712 was increased in the range of 19–36, 21–35, 18–79 and 18–55 %, respectively by NaCl treatments. Increased CAT activity of Pusa-37 genotype, and no significant increase in the CAT activity of its contemporary genotypes (BRAGG and PK-1042) in response to NaCl treatments grant additional salt tolerance in the former genotype. Neto et al. [33] also found higher CAT activity in two maize cultivars differing in salt tolerance. A number of other reports are available on the effect of salt stress on CAT activity in several plant species. CAT activity has been found to increase under salt stress in soybean [34], tobacco [35], cucumber [36] and mulberry [37].
The first enzyme of the ascorbate–glutathione cycle, APX, catalyses the reduction of H2O2 to water and has high specificity and affinity for ascorbate as reductant [38]. It has been reported that APX activity may have an important role in the mechanism of salt tolerance in plants. The APX activity was significantly higher in Pusa-37 (∼5-fold), followed by BRAGG and PK-1042 at 150 mM NaCl treatment. Gueta-Dahan et al. [39] indicated that acquisition of salt tolerance might also be a consequence of improving resistance to salt stress, via increased APX activity. Increased activity of APX in salt-adapted cells seems to be more important for their salt tolerance. Bor et al. [40] found induced APX activity in salt-tolerant wild beet. Mittova et al. [41] also reported same relation between APX and salt tolerance in tomato. There was no significant change in the APX activities in PK-416 and Pusa-20 by NaCl treatments, compared with control. APX activity had a key role in response to salt stress in the comparison of the activities of antioxidant enzymes in salt-sensitive and salt-tolerant citrus cells [39].
GR is the last enzyme of ascorbate–glutathione cycle and catalyses the NADPH-dependant reduction of GSSG. APX reduces H2O2 into water using ascorbate as an electron donor, resulting in the formation of dehydroascorbate. It is recycled back to ascorbate using reduced glutathione (GSH) as an electron donor, and the GSSG is converted back to GSH by NADPH-dependent GR [42]. Therefore, GR is one of the three enzymes, which catalyse reaction that maintain large pool of GSH and ascorbate in the H2O2 scavenging pathway in chloroplasts. The role of GR in antioxidative defence mechanism under NaCl stress is observed in present study. Enhancement in GR activity due to increasing salinity seemed to be a common response in all the soybean genotypes. However, with the highest level of NaCl stress Pusa-37 showed the maximum GR activity while PK-416 showed the minimum. The increased GR activity is in correlation with the increase in APX activity. Hence, the growth of this genotype was less influenced by salinity. There are reports showing that GR activity increased in NaCl-tolerant pea variety as compared with NaCl-sensitive pea [23].
Proline accumulation is one of the most frequently reported modifications induced by salt stress in plants and is often considered to be involved in stress resistance mechanisms. Cytoplasmic accumulation of this amino acid is thought to be involved in osmotic adjustment of stressed tissues [43, 44]. Proline is also considered to be involved in the protection of enzymes [45] and cellular structures [46] and to act as a free radical scavenger [47]. Finally, its biosynthesis could be associated with the regulation of cytosolic pH [48] or with the production of NADP for the stimulation of the pentose phosphate pathway [49]. Sharp increases in proline levels were reported under the effect of salinity. Proline accumulation in Pusa-16, Pusa-22, Pusa-40 and DS-9712 found highest at 75 mM NaCl, while significant change in this osmoprotectant was observed over 25 mM NaCl in PK-416 and Pusa-20. In our study, we observed increase in proline content of Pusa-37 (1.8-fold) with increased NaCl dose, followed by BRAGG and PK-1042. Changes in the proline content due to NaCl stress suggest that permeability of membranes might be affected as it was observed earlier [50]. In salt-tolerant plants, Beta vulgaris [51], Brassica juncea [29] and alfalafa [52], sharp increases in proline levels were reported under the effect of salinity.
In the present study, proteomic analysis in salt-tolerant genotype (Pusa-37) led to the detection of proteins involved in a variety of biological processes, such as protein synthesis (12 %), redox regulation (19 %), primary and secondary metabolism (25 %), or disease- and defence-related processes (32 %) (Fig. 9). Similar observations were observed by Witzel et al. [54] in the root proteome of barley genotypes. A total of 173 protein spots were detected; more than 40 proteins were differentially expressed by salt stress, and 16 of these were structurally analysed. The relative intensity of four proteins increased by 2- to 7-fold and that of three proteins decreased by 4- to 6-fold in salt-stressed plants as compared with the untreated control. Glycine dehydrogenase (decarboxylating, 40) was found to be lightly upregulated in response to salinity. It is the enzyme which is involved in the C-2 glycolate pathway in higher plants that can decarboxylate glycine to form serine, which can generate ethanolamine after decarboxylation again. Moreover, ethanolamine can undergo a series of methylations to form choline and then glycine betaine is formed as a result of the oxidation of choline. We found that photosystem II oxygen-evolving enhancer protein (spot 33) precursor was upregulated in response to salt stress. This is in conformity with the observations of Abbasi and Komatsu [55], which showed that this protein was upregulated in rice leaf sheath under salt stress. Riblose large subunit and riblose-5-phosphate isomerase (spots 12 and 30) are the key enzyme of the Calvin cycle. It was noted that the abundance of riblose large subunit decreased in response to salinity. Spot No. 10 was identified as UDP-glucose pyrophosphate. UGPase is responsible for the synthesis and pyrophosphorolysis of UDP-glucose, the key precursor of sucrose and cell wall components (e.g. cellulose and glucans). In Arabidopsis, it was strongly induced by sucrose, light, cold stress and phosphate deficiency but reduced by drought and flooding [6, 23]. Our observation confirms the results of Yan et al. [56] that the UGPase protein gets markedly reduced by salt stress. Putative monodehydroascorbate reductase antioxidant protein (spot 28) was the first peroxiredoxin to be identified. The peroxiredoxins are a family of multiple isozymes that catalyse the reduction of H2O2 and protect the cells against oxidative damage in plants. The upregulation of the thiol-specific antioxidant protein, identified in this study, indicates that it might play an important role in ROS scavenging under salt stress. Ferrodoxin-like protein (spots 27) is a flavoenzyme that catalyses the reversible electron transfer between NADPH and (2Fe–2S) ferredoxins or flavodoxins [52]. In plants ferredoxin, NADP+ oxidoreductase is implicated in photosynthesis and nitrogen fixation in plastids; recently, a new role was proposed for it in cell protection against ROS, but at present, the mechanism by which ferredoxin NADP+ oxidoreductase protects cells against the presence of ROS in plants remains unknown [20]. In wheat, salt stress induced an upregulation of ferredoxin NADP+ oxidoreductase. CAT (spot No. 14) was found to be over-expressed under salinity stress. It is the main scavenger of H2O2 in peroxisomes, converting it to water and molecular oxygen [32]. Among the identified spots, the proteins whose level of expression is higher include MDAR, two spots of different molecular weight identified as l-APX, and lactoylglutathione lyase, which was identified in a second spot with a higher molecular weight as compared with the protein spot in the first group. The induction of APX by salt-stress treatment was not unexpected since it catalyses the reduction of H2O2 to water with the concomitant generation of monodehydroascorbate and is therefore a key component in the scavenging pathway of ROS produced by various stress factors [57]. The accumulation of APX upon salinity treatment is in agreement with the literature [57–60].

Assignment of the identified proteins to functional categories using the classification described by Bevan et al. [53]. A total of 40 spots representing 16 different proteins were classified. If a spot contained two proteins, it was counted twice
Conclusions
In conclusion, the soybean plants in our study responded to salt stress by changing their protein expression pattern. This study gives new insights into the salt-stress response in soybean and demonstrates the power of the proteomic approach in plant biology studies which could finally help us in identifying the possible regulatory switches (gene/s) controlling novel proteins of the salt-tolerant genotype of the crop plants and their possible role in defence mechanism.
Abbreviations
- APX:
-
Ascorbate peroxidase
- BSA:
-
Bovine serum albumin
- CAT:
-
Catalase
- DW:
-
Dry weight
- EDTA:
-
Ethylenediaminetetraacetic
- FW:
-
Fresh weight
- GR:
-
Glutathione reductase
- GSH:
-
Reduced glutathione
- GSSG:
-
Oxidised glutathione
- MDA:
-
Malondialdehyde
- NBT:
-
Nitroblue tetrazolium
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- TBA:
-
Thiobarbituric acid
- TCA:
-
Trichloroacetic acid
- MALDI-TOF-MS:
-
Matrix-assisted laser desorption ionisation time-of-flight mass spectrometry
References
FAO. (2005). Global network on integrated soil management for sustainable use of salt-affected soils. Rome: FAO land and plant nutrition management service.
Nemoto, Y., & Sasakuma, T. (2002). Differential stress responses of early salt-stress responding genes in common wheat. Phytochemistry, 61, 129–133.
Locy, R. D., Chang, C. C., Nielsen, B. L. & Singh, N. K. (1996). Photosynthesis in salt-adapted heterotrophic tobacco cells and regenerated plants. Plant Physiology, 110, 321–328.
Singh, R. K., Ghosh, P. K., Bandyopadhyay, K. K., Misra, A. K., Mandal, K.G. & Hati, K. M. (2006). Integrated plant nutrient supply for sustainable production in soybean – based cropping system. Indian Journal of Fertilizers 1(11), 25–32.
Hoagland, D. R., & Arnon, D. S. (1950). The water culture method for growing plants without soil. California Agricultural Experiment Station Circular, 347, 1–32.
Heath, R. L., & Packer, L. (1968). Photoperoxidation in isolated chloroplasts I. Kinetic and stoichiometry of fatty acid peroxidation. Archives of Biochemistry and Biophysics, 125, 189–198.
Bates, L. S., Walden, R. P., & Teare, I. D. (1973). Rapid determination of free proline for water-stress studies. Plant and Soil, 39, 205–207.
Beauchamp, C. O., & Fridovich, I. (1971). Superoxide dismutase: improved assays and assay applicable to acrylamide gels. Analytical Biochemistry, 44, 276–287.
Giannopolitis, C. N., & Ries, S. K. (1977). Superoxide dismutase. I. Occurrence in higher plants. Plant Physiology, 59, 309–314.
Nakano, Y., & Asada, K. (1981). Hydrogen peroxide is scavenged by ascorbate specific peroxidase in spinach chloroplasts. Plant and Cell Physiology, 22, 867–880.
Aebi, H. (1984). Catalase in vitro. Methods in Enzymology, 105, 121–126.
Foyer, C. H., & Halliwell, B. (1976). The presence of glutathione and glutathione reductase in chloroplasts: a proposed role in ascorbic acid metabolism. Planta, 133, 21–25.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Damerval, C., Vienne, D. D., Zivy, M. & Thiellement, H. (1986). Technical improvements in two-dimensional electrophoresis increase the level of genetic variation detected in wheat-seedling proteins. Electrophoresis, 7, 52–54.
O’Farrell, P. H. (1975). High resolution two-dimensional electrophoresis of proteins. Journal of Biological Chemistry, 250, 4007–4021.
Chivasa, S., Ndimba, B. K., Simon, W. J., Robertson, D., Yu, X. L., Knox, J. P., Bolwell, P. & Slabas, A. R. (2002). Proteomic analysis of the Arabidopsis thaliana cell wall. Electrophoresis, 23, 1754–1765.
Laemilli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227 (5259), 680–685.
FAO (2000). Global network on integrated soil management for sustainable use of salt affected soils. Available from: http://www.fao.org/ag/AGL/agll/spush/intro.htm.
Munns, R. (2000). Comparative physiology of salt and water stress. Plant Cell and Environment, 25, 239–250.
Ruiz, J. M., Blasco, B., Rivero, R. M., & Romero, L. (2005). Nicotine-free and salt tolerant tobacco plants obtained by grafting to salinity-resistant root stocks of tomato. Physiologia Plantarum, 124, 465–475.
Greenway, H., & Munns, R. (1980). Mechanisms of salt tolerance in non-halophytes. Annual Reviews of Plant Physiology, 31, 149–190.
Gomez, J. M., Hernendez, J. A., Jimenez, A., del Rio, L. A., & Sevilla, F. (1999). Differential response of antioxidative enzymes of chloroplasts and mitochondria to long term NaCl stress of pea plants. Free Radical Research, 31, S11–S18.
Hernandez, J. A., Jimenez, A., Mullineaux, P., & Sevilla, F. (2000). Tolerance of pea (Pisum sativum L.) to a long term salt stress is associated with induction of antioxidant defences. Plant Cell and Environment, 23, 853–862.
Demiral, T., & Turkan, I. (2005). Comparative lipid peroxidation, antioxidant defense systems and proline content in roots of two rice cultivars differing in salt tolerance. Environmental and Experimental Botany, 53, 247–257.
Khan, M. H., Singh, L. B., & Panda, S. K. (2002). Changes in antioxidant levels in Oryza sativa L. roots subjected to NaCl-salinity stress. Acta Physiologia Plant, 24, 145–148.
Mandhania, S., Madan, S., & Sawhney, V. (2006). Antioxidant defense mechanism under salt stress in wheat seedlings. Biologia Plantarum, 227, 227–231.
Panda, S. K., & Khan, M. H. (2003). Salt stress influences lipid peroxidation and antioxidants in the leaf of an indica rice (Oryza sativa L). Physiology and Molecular Biology of Plants, 9, 273–278.
Panda, S. K., & Upadhyay, R. K. (2003). Salt stress injury induces oxidative alteration and antioxidative defense in the roots of Lemna minor. Biologia Plantarum, 48, 249–253.
Jain, M., Mathur, G., Koul, S., & Sarin, N. B. (2001). Ameliorative effects of proline on salt stress-induced lipid peroxidation in cell lines of groundnut (Arachis hypogaea L.). Plant Cell Reports, 20, 463–468.
Fadzilla, N. M., Finch, R. P., & Burdon, R. H. (1997). Salinity, oxidative stress and antioxidant responses in shoot cultures of rice. Journal of Experimental Botany, 48, 325–331.
Sairam, R. K., Srivastava, G. C., Agarwal, S., & Meena, R. C. (2005). Differences in antioxidant activity in response to salinity stress in tolerant and susceptible wheat genotypes. Biolgia Plantarum, 49, 85–91.
Willekens, H., Inze, D., Van Montagu, M., & Van Camp, W. (1995). Catalase in plants. Molecular Breeding, 1, 207–228.
Neto, A. D. A., Prisco, J. T., Eneás-Filho, J., Abreu, C. E. B., & Gomes-Filho, E. (2006). Effect of salt stress on antioxidative enzymes and lipid peroxidation in leaves and roots of salt-tolerant and salt-sensitive maize genotypes. Environmental and Experimental Botany, 56, 87–94.
Comba, M. E., Benavides, M. P., Gallego, S. M., & Tomaro, M. L. (1998). Relationship between nitrogen fixation and oxidative stress induction in nodules of salt treated soybean plants. Phyton-International Journal of Experimental Botany, 60, 115–126.
Bueno, P., Piqueras, A., Kurepa, J., Savoure, A., Verbruggen, N., Montagu, V. M., et al. (1998). Expression of antioxidant enzymes in response to abscisic acid and high osmoticum in tobacco BY-2 cell cultures. Plant Science, 138, 27–34.
Lechno, S., Zamski, E., & Tel-Or, E. (1997). Salt stress-induce responses in cucumber plants. Journal of Plant Physiology, 150, 206–211.
Sudhakar, C., Lakshmi, A., & Giridarakumar, S. (2001). Changes in the antioxidant enzyme efficacy in two high yielding genotypes of mulberry (Morus alba L.) under NaCl salinity. Plant Science, 161, 613–619.
Asada, K. (1999). The water–water cycle in chloroplasts: scavenging of active oxygen and dissipation of excess photons. Annual Reviews of Plant Physiology and Plant Molecular Biology, 50, 601–639.
Gueta-Dahan, Y., Yaniv, Z., Zilinskas, B. A., & Ben-Hayyim, G. (1997). Salt and oxidative stress: similar and specific responses and their relation to salt tolerance in citrus. Planta, 203, 460–469.
Bor, M., Özdemir, F., & Türkan, I. (2003). The effect of salt stress on lipid peroxidation and antioxidants in leaves of sugar beet Beta vulgaris L. and wild beet Beta maritima L. Plant Science, 164, 77–84.
Mittova, V., Volokita, M., Guy, M., & Tal, M. (2000). Activities of SOD and the ascorbate–glutathione cycle enzymes in subcellular compartments in leaves and roots of the cultivated tomato and its wild salt-tolerant relative Lycopersicon pennilli. Physiologia Plantarum, 110, 42–51.
Asada, K., & Takahashi, M. (1987). Photoinhibition. In D. J. Kyle, C. B. Osmond, & C. J. Arntzen (Eds.), Production and scavenging of active oxygen in photosynthesis (pp. 227–287). Amsterdam: Elsevier.
Delauney, A. J., & Verma, D. P. S. (1993). Proline biosynthesis and osmoregulation in plants. The Plant Journal, 4, 215–223.
Kavi-Kishor, P. B., Hong, Z., Miao, G. H., Hu, C. A. A., & Verma, D. P. S. (1995). Overexpression of D1-pyrroline-5-carboxylate synthetase increases proline production and confers osmotolerance in transgenic plants. Plant Physiolgy, 108, 1387–1394.
Solomon, A., Beer, S., Waisel, Y., Jones, G. P., & Paleg, L. G. (1994). Effects of NaCl on the carboxylating activity of Rubisco from Tamarix jordanis in the presence and absence of proline-related compatible solutes. Physiologia Plantarum, 90, 198–204.
Van Rensburg, L., Kruger, G. H. J., & Kruger, H. (1993). Proline accumulation as drought-tolerance selection criterion: its relationship to membrane integrity and chloroplast ultrastructure in Nicotiana tabacum L. Journal of Plant Physiology, 141, 188–194.
Alia, Prasad, K. V. S. K., & Pardha-Saradhi, P. (1995). Effect of zinc on free radicals and proline in Brassica and Cajanus. Phytochemistry, 39, 45–47.
Venekamp, J. H. (1989). Regulation of cytosol acidity in plants under conditions of drought. Physiologia Plantarum, 70, 381–388.
Hagedorn, C. H., & Phang, J. M. (1986). Catalytic transfer of hydride ions from NADPH to oxygen by the inter conversions of proline and delta1-pyrroline-5-carboxylate. Archives of Biochemistry and Biophysics, 248, 166–174.
Ali, G., Srivastava, P. S., & Iqbal, M. (1999). Proline accumulation, protein pattern and photosynthesis in regenerants grown under NaCl stress. Biologia Plantarum, 42, 89–95.
Gzik, A. (1996). Accumulation of proline and pattern of amino acids in sugar beet plants in response to osmotic, water and salt stress. Environmental and Experimental Botany, 36, 29–38.
Petrusa, L., & Winicov, I. (1997). Proline status in salt tolerant and salt sensitive alfalfa cell lines and plants in response to NaCl. Plant Physiology and Biochemistry, 35, 303–310.
Bevan, M., Bancroft, I., Bent, E., Love, K., Goodman, H., Dean, C., et al. (1998). Analysis of 1.9Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature, 391, 485–488.
Witzel, K., Weidner, A., Surabhi, G. K., Börner, A. & Mock, H. P. (2009). Salt stress-induced alterations in the root proteome of barley genotypes with contrasting response towards salinity. Journal of Experimental Botany, 60, 3545–3557.
Abbasi, F. M., & Komatsu, S. (2004). A proteomic approach to analyze salt responsive proteins in rice leaf sheath. Proteomics, 4, 2072–2081.
Yan, S., Tang Z., Su, W. & Sun, W. (2005). Proteomic analysis of salt stress-responsive proteins in rice root. Proteomics, 5, 235–244
Shigeoka, S., Ishikawa, T., Tamoi, M., Miyagawa, Y., Takeda, T., Yabuta, Y., et al. (2002). Regulation and function of ascorbate peroxidase isoenzymes. Journal of Experimental Botany, 53, 1305–1319.
Jiang, Y., Yang, B., Harris, N. S. & Deyholos, M. K. (2007). Comparative proteomic analysis of NaCl stress-responsive proteins in Arabidopsis roots. Journal of Experimental Botany, 58, 3591–3607.
Caruso, G., Cavaliere, C., Guarino, C., Gubbiotti, R., Foglia, P. & Laganà, A. (2008). Identification of changes in Triticum durum L. leaf proteome in response to salt stress by two-dimensional electrophoresis and MALDI-TOF mass spectrometry. Analytical and Bioanalytical Chemistry, 391,381–390.
Wang, M. C., Peng, Z. Y., Li, C. L., Li, F., Liu, C. & Xia, G. M. (2008). Proteomic analysis on a high salt tolerance introgression strain of Triticum aestivum/Thinopyrum ponticum. Proteomics, 8, 1470–1489.
Author information
Authors and Affiliations
Corresponding author
Additional information
Khalid Rehman Hakeem, Faheema Khan and Ruby Chandna contributed equally to this work.
Rights and permissions
About this article
Cite this article
Hakeem, K.R., Khan, F., Chandna, R. et al. Genotypic Variability Among Soybean Genotypes Under NaCl Stress and Proteome Analysis of Salt-Tolerant Genotype. Appl Biochem Biotechnol 168, 2309–2329 (2012). https://doi.org/10.1007/s12010-012-9939-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-012-9939-6