
Abstract
A psychrotrophic Pseudomonas sp. TK-3 was isolated from dirty and cool stream water in Toyama, Japan from which we cloned and characterized the bacterial lipase LipTK-3. The sequenced DNA fragment contains an open reading frame of 1,428 bp that encoded a protein of 476 amino acids with an estimated molecular mass of 50,132 Da. The lipase showed high sequence similarity to those of subfamily Ι.3 lipase and had a conserved GXSXG motif around the catalytic Ser residue. Its optimal temperature was 20–25 °C, lower than in most other subfamily Ι.3 lipases. The lipase exhibited about 30 % of maximal activity at 5 °C. The optimal pH value was 8.0. The activity was strongly inhibited by EDTA and was highly dependent on Ca2+. Tricaprylin and p-nitrophenyl caprylate were the most favorable substrates among the triglycerides and p-nitrophenyl esters, respectively. LipTK-3 also showed high activity towards natural substrates including edible vegetable oils and animal fats. Furthermore, LipTK-3 was very active and stable in the presence of several detergents, metal ions, and organic solvents. This cold-adapted lipase may prove useful for future applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lipases (triacylglycerol acylhydrolases, EC3.1.1.3) catalyze the hydrolysis of triacylglycerides at the interface between the insoluble substrate and water [1]. In general, lipases present a typical α/β hydrolase fold and a catalytic triad consisting of three residues: serine, aspartate or glutamate, and histidine [2]. In addition, they have a well-conserved GXSXG pentapeptide motif where the active site serine residue lies. They are ubiquitous in nature, and are produced by various plants, animals, and microorganisms. Lipases of microbial origin are widely diversified in their catalytic activities and substrate specificities, which make them attractive tools for industrial applications.
Owing to their useful features, microbial lipases are used in a wide array of industrial applications, such as food technology, detergents, biodiesel, chemical industry, and biomedical sciences [3, 4]. Several bacterial lipases have been purified and characterized, and their corresponding genes have been cloned and sequenced [5–11]. Bacterial lipolytic enzymes have been classified into eight families, I to VIII, according to their amino acid sequence and biological properties [12]. Lipases in family I can be classified into seven subfamilies, subfamilies I.1 to I.7. Most of the lipases from the genus Pseudomonas fall in three subfamilies, I.1, I.2, and I.3 [1, 3, 12]. Although the lipases of subfamilies I.1 and I.2 are clearly homologous with amino acid sequence identities above 30 %, the subfamily I.3 enzymes exhibit only a very low sequence similarity to the former two subfamilies. Unlike the well-studied subfamily I.1 and I.2 lipases, there is relatively less information on the structures and functions of subfamily I.3 lipases [13]. Subfamily I.3 lipases have molecular masses of 50–68 kDa and do not have cysteine residues. The C-terminal domains of these proteins contain the secretion signal, and several repeats of the GGXGXDXUX sequence motif (X: any amino acid; U: hydrophobic amino acid) that form a β-roll motif in the presence of Ca2+ [13]. Subfamily I.3 lipases are secreted via the type I secretion system (T1SS) also called the ATP-binding cassette (ABC) transporter system [13, 14].
In particular, cold-adapted lipases, which display high lipolytic activity at low temperatures, are attractive biocatalysts for biotechnology applications. For instance, they have great potential in production of pharmaceuticals and cosmetics, food industry, synthesis of biodiesel, wastewater treatment, bioremediation in fat-contaminated cold environments, and additives in detergents for cold washing [15, 16]. Psychrophillic and psychrotrophic microorganisms could be good candidates to produce these types of lipases. So far, the cold-adapted microorganisms have been studied, and their potential applications have been examined [5–9, 11, 16].
In the present study, a psychrotrophic Pseudomonas sp. strain, designated as TK-3, was isolated from dirty and cool stream water. Furthermore, we cloned and characterized the subfamily I.3 lipase from the bacterial strain to assess its potential for industrial applications.
Materials and Methods
Isolation of lipase-producing Bacteria
Microorganisms were isolated from dirty and cool water samples collected from several different streams in a residential area of Toyama, Japan. For the first screening of lipase-producing microorganisms, a small volume of each sample was spread on standard method agar (Nissui Pharmaceutical Company, Tokyo, Japan) containing 1 % (w/v) canola oil and cultivated at 25 °C. The bacterial colonies that formed transparent zones were isolated. One strain, designated as TK-3, showed the highest lipolytic activity at 5 °C and 25 °C and was selected for further study.
Bacterial Identification
The morphology and motility of the isolated bacteria were initially observed under a phase contrast microscope (BX51; Olympus Optical Co. Ltd., Tokyo, Japan). The cells were then negatively stained with 1 % (w/v) phosphotungstic acid and observed with a JEM-1200EXII electron microscope (JEOL, Tokyo, Japan). Micrographs were taken at an accelerating voltage of 80 kV. Biochemical characterization was performed using the API 20NE (BioMerieux, Marcy, l'Etoile, France). In addition, gram staining (Favor G set, Nissui Pharmaceutical, Tokyo, Japan), catalase test, and oxidase test were conducted.
For 16S rRNA gene sequencing, bacterial colonies were picked and suspended in 100 μl of sterile water. Each suspension was heated to 100 °C for 10 min to release DNA into the water. Almost the full length of the bacterial 16S rRNA gene was amplified by PCR using the primers 27f (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1525r (5′-AAAGGAGGTGATCCAGCC-3′). The amplified products were purified using a QIAquick PCR purification kit (Qiagen, Tokyo, Japan) and sequenced directly using the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) and an ABI Prism 3130xl genetic analyzer (Applied Biosystems). The DNA sequence was searched by the Basic Local Alignment Search Tool (BLAST) program on the NCBI website (http://www.ncbi.nlm.nih.gov). A phylogenetic tree was constructed using the neighbor joining method with Kimura 2 parameter distances in MEGA version 5.0 software. The dataset was bootstrapped 1,000 times.
Cloning and Sequencing of the Lipase Gene
Genomic DNA was isolated using phenol-chloroform extraction and partially digested with Sau3AI (Nippon Gene, Tokyo, Japan). The resulting DNA fragments were separated by agarose gel electrophoresis, and 3–10 kbp DNA fragments were recovered and ligated with pUC19, which had been previously digested with BamHI (Nippon Gene) and dephosphorylated with bacterial alkaline phosphatase. The ligation products were introduced into Escherichia coli DH5α (Nippon Gene). To screen for lipase activity, the transformants were grown on Luria–Bertani (LB) agar (1 % tryptone, 0.5 % yeast extract, 1 % NaCl, and 1.5 % agar) supplemented with 1 % (w/v) tributyrin, ampicillin (50 μg ml−1), isopropyl-β-d-thiogalactopyranoside (IPTG) (50 μl of 0.1 M stock per plate), and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) (50 μl of 4 % stock per plate). White colonies with clear halos were then selected, and recombinant plasmids were extracted using a PureYield Plasmid Miniprep System (Promega, Madison, WI, USA). The inserted DNA fragments were amplified by PCR using universal M13 primers targeting both ends of the cloning vector and DNA sequencing of the inserted DNA fragments was performed as described above. Nucleotide and deduced amino acid sequence analyses, open reading frame search, multiple alignment, and molecular-mass and isoelectric-point calculations were performed using Genetyx Ver.8 software (Genetyx, Tokyo, Japan). A database homology search was performed with the BLAST program on the NCBI website.
Overexpression and Purification of Recombinant Lipase LipTK-3
The open reading frame of the putative lipase gene was amplified by PCR using the primers 5′-CTCTTTCAGGGACCCATGGGTATCTTTGACTA-3′ and 5′-CTGCGGCCGCAAGCTTTCAGCCGATGCTGACG-3′. The PCR fragments were cloned between the SmaI and HindIII sites of pET-47b expression vector (Novagen, Madison, WI, USA) using the In-Fusion Advantage PCR Cloning Kit (Takara Bio, Shiga, Japan). This recombinant plasmid, designated as pET-lip, was used for the expression of the lipase gene in E. coli BL21 (DE3). No changes were observed in the nucleotide sequence of the lipase gene after cloning. E. coli cells carrying pET-lip was grown overnight at 37 °C with shaking in LB broth containing kanamycin (25 μg ml−1). The preculture was inoculated (1 %) into fresh LB broth containing kanamycin, and cultivation was continued until the optical density at 550 nm reached 0.4. The culture was then supplemented with 0.5 mM (final concentration) isopropyl-β-d-thiogalactopyranoside (IPTG) and incubated for another 8 h at 37 °C for overexpression of the lipase gene. Cells were harvested by centrifugation at 10,000×g for 10 min and resuspended in BugBuster protein extraction reagent (Novagen) at 5 ml BugBuster per gram wet-cell pellet and kept on ice for an additional 15 min. Soluble and insoluble fractions were separated by centrifugation (16,000×g for 20 min). The recombinant lipase in the insoluble form was denatured with 6 M urea and then refolded by fractional dialysis in 3, 1.5, and 0 M urea in 50 mM Tris–HCl (pH 8.0). The refolded protein was purified by a His GraviTrap column (GE Healthcare, UK). The purity of the protein was examined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). SDS-PAGE was performed according to Laemmli [17] using 10 % acrylamide gels. Proteins were visualized in the gels by staining with Coomassie brilliant blue. The lipase activity of the enzyme protein band was confirmed by zymogram analysis [18]. After electrophoresis, SDS was removed by washing the gel once for 20 min with 20 % isopropanol and then twice for 10 min with distilled water. For lipase activity detection, the gel was transferred to a 1.3 % agar plate containing 1 % tributyrin, 25 mM Tris–HCl (pH 8.0), and 5 mM CaCl2. After incubation for 6 h at 25 °C, bands of lipase activity were visualized as clearing bands of tributyrin hydrolysis. Protein concentrations were determined using the Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL, USA) with bovine serum albumin as a standard.
Lipase Assay
The lipase activity assay using p-nitrophenyl esters was performed as described previously [19, 20], with some modifications. The reaction mixture consisted of 165 μl of 0.4 % (w/v) Triton X-100, 0.1 % (w/v) gum arabic in 50 mM Tris–HCl buffer (pH 8.0), 15 μl of substrate (16.6 mM p-nitrophenyl ester in isopropanol), and 10 μl of 100 mM CaCl2. The mixture was pre-incubated at 25 °C for 10 min, and 10 μl of enzyme solution was subsequently added. The enzyme solution contained purified lipase in 50 mM Tris–HCl buffer (pH 8.0) at a concentration of 0.2 μg ml−1. After 20 min of incubation at 25 °C, the color change was measured immediately at 405 nm using a microplate reader (Model 550, Bio-Rad, CA, USA). One unit (U) of lipase activity was defined as the amount of enzyme needed to release 1 μmol of p-nitrophenol per minute.
Lipase activity was also examined by titrating free fatty acids from triglycerides as described previously [10], with some modifications. The reaction mixture consisted of 1.8 ml of 0.4 % (w/v) Triton X-100, 0.1 % (w/v) gum arabic in 50 mM Tris–HCl buffer (pH 8.0), 0.3 g of triglyceride, and 100 μl of 100 mM CaCl2. The mixture was pre-incubated at 25 °C for 10 min, and 100 μl of enzyme solution was subsequently added. The enzyme solution contained purified lipase in 50 mM Tris–HCl buffer (pH 8.0) at a concentration of 0.2 μg ml−1. After 20 min of incubation at 25 °C with shaking at 170 rpm, the reaction was terminated by the addition of 7 ml of acetone–ethanol (1:1, v/v) and the liberated fatty acid was titrated with 10 mM NaOH. One unit of lipase activity was defined as the amount of enzyme that liberated 1 μmol of fatty acid per minute.
Characterization of Lipase
The p-nitrophenyl caprylate (C8) was subsequently used as a substrate to assay the enzyme's characteristics. The optimum temperature for the lipase activity of LipTK-3 was determined by assaying for its hydrolytic activity at various temperatures in 50 mM Tris–HCl buffer, pH 8.0. To examine its thermostability, the enzyme was incubated at various temperatures for 1 h in 50 mM Tris–HCl buffer (pH 8.0), and the residual activity was then determined at 20 °C and pH 8.0. The optimum pH was determined between 4 and 10 at 25 °C. The buffer used was GTA buffer, which is an universal buffer consisting of 50 mM 3,3-dimethylglutaric acid, 50 mM Tris, and 50 mM 2-amino-2-methyl-1,3-propanediol. The effects of metal ions and detergents on LipTK-3 were analyzed by measuring the activities in 50 mM Tris–HCl buffer (pH 8.0) containing 1 and 5 mM of various metal ions and 0.005 % (w/v) detergents, respectively. The stabilities of LipTK-3 towards organic solvents were surveyed by determining the residual activity after 1 h incubation at 25 °C in 50 mM Tris–HCl buffer (pH 8.0) containing 20 % (v/v) of various organic solvents.
Nucleotide Sequence Accession Numbers
The nucleotide sequences of the 16S rRNA gene and lipase gene determined in this study have been deposited in GenBank/EMBL/DDBJ under the accession numbers AB705621 and AB705622, respectively.
Results
Identification of Strain TK-3
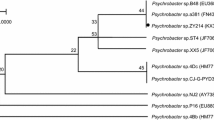
A strain, named TK-3, showed a clear halo when grown at 5 °C on an agar plate containing 1 % canola oil. Growth occurred at temperatures ranging from −5 to 35 °C, with an optimum at 25 °C. This strain was Gram-negative, rod-shaped, motile by means of one to three polar flagella (Fig. 1). It was also shown to be aerobic, catalase-positive, and oxidase-positive. From the API 20 NE results, this strain was shown to have a 96.9 % ID match with Pseudomonas fluorescens. The 16S rRNA gene sequence showed high sequence similarity to Pseudomonas species (Fig. 2), the highest degree of similarity attained was 99.9 % with Pseudomonas sp. W15Feb2 (EU680980). Thus, strain TK-3 was identified as a member of the genus Pseudomonas.

Transmission electron micrograph of a negatively stained cell of Pseudomonas sp. TK-3; bar, 1 μm

Phylogenetic analysis of the 16S rRNA gene sequences of strain TK-3 within the genus Pseudomonas by the neighbor-joining method. The accession numbers of the sequences are in parentheses. Bootstrap values (percent) are indicated at the nodes; only values greater than 50 are shown. The scale bar represents 0.005 substitutions per base position
Gene Cloning, Expression, and Purification of LipTK-3
The lipase gene from Pseudomonas sp. TK-3 was cloned into E. coli DH5α using the shotgun method. About 10,000 transformants carrying recombinant plasmids were screened using agar plates containing 1 % tributyrin, and two lipase-producing E. coli transformants were detected. The plasmids prepared from the clones had an identical insert of 3 kbp. Sequence analysis identified a lipase ORF of 1,428 bp that encoded a protein of 476 amino acids with an estimated molecular mass of 50,132 Da, isoelectric point of 4.8, and G + C content of 61 %. The deduced amino acid sequences of the ORF had no cysteine residue and showed high similarity with lipases classified in subfamily I.3. A BLAST homology search indicated that it shared amino acid sequence identity of 96 % to P. fluorescens B52 lipase (LipB52; AAA25882), 95 % to P. fluorescens B68 lipase (LipB68; AAU10321), 91 % to Pseudomonas sp. KB700A lipase (KB-Lip; BAB64913), 91 % to P. fluorescens SIK W1 lipase (TliA; AAD09856), 80 % to Pseudomonas sp. 7323 lipase (LipA; CAJ76166), 79 % to Pseudomonas sp. MIS38 lipase (PML; BAA84997), and 78 % to P. fluorescens HU380 lipase (LipA; BAC98496). TK-3 lipase contained the catalytic triad (Ser207, Asp255, and His313) and the conserved GXSXG motif around the catalytic Ser residue (Fig. 3). TK-3 lipase did not contain an N-terminal signal sequence, while its C-terminal region contained five repeats of a nine-residue GGXGXDXUX sequence motif and two putative secretion signals. These data indicate that its secretion mechanism might follow the T1SS [13].

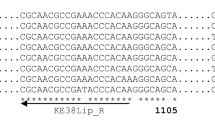
Alignment of the amino acid sequence of lipase from Pseudomonas sp. TK-3 (TK-3) with homologous lipases from P. fluorescens B52 (B52), P. fluorescens B68 (B68), Pseudomonas sp. KB700A (KB700A), and P. fluorescens SIK W1 (SIK W1). The identical and conserved residues are marked by asterisk and dot, respectively. The consensus GXSXG sequence, containing the active-site serine residue, is shaded. The amino acid residues forming a catalytic triad, Ser, Asp, and His, are indicated by solid circles. The repetitive nine residue sequence motif, GGXGXDXUX, is boxed by a solid line. Two putative secretion signals located at the C terminus are underlined
LipTK-3 was highly expressed as an N-terminal His-tag fusion protein in E. coli BL21 (DE3) containing pET-lip, and the recombinant lipase protein was about 50 kDa and demonstrated lipolytic activity on the overlaid tributyrin agar plate (Fig. 4). The recombinant lipase protein was purified to greater than 90 % homogeneity as judged by densitometric scanning of SDS-PAGE gels.

SDS-PAGE (a) and zymogram (b) analysis of recombinant lipase LipTK-3. Lane M, molecular weight marker; lane 1, crude extract; lane 2, purified lipase
Enzyme Characterization
The purified LipTK-3 was most active at pH 8.0. Therefore, we examined the effects of temperature on the activity of LipTK-3 at pH 8.0. LipTK-3 exhibited maximum activity between 20 and 25 °C and about 30 % of maximal activity at 5 °C (Fig. 5). The residual activity was about 40 % after treatment at 30 °C for 1 h and was abolished after treatment at 60 °C for 1 h.

Effect of temperature on activity (solid circle) and stability (solid square) of LipTK-3. The enzyme activity was assayed with p-nitrophenyl caprylate as the substrate at various temperatures for 20 min. For thermal stability, the enzyme was assayed after incubation at various temperatures for 1 h
The lipase activities of the purified LipTK-3 were examined using various triglycerides, p-nitrophenyl esters, and natural oils at 25 °C and pH 8.0 (Table 1). Results showed that tricaprylin (C8:0) and p-nitrophenyl caprylate (C8) were the most favorable substrates among the triglycerides and p-nitrophenyl esters, respectively. The specific activities towards tricaprylin and p-nitrophenyl caprylate were 746.4 and 329.4 U/mg, respectively. Furthermore, LipTK-3 showed high activity towards natural substrates such as sesame oil, soybean oil, and lard.
The effects of metal ions on the activity of LipTK-3 were examined (Table 2). Lipase activity was inhibited by Zn2+, Cd2+, Pb2+, and Cu2+ even at the lowest tested concentration (1 mM). Complete inactivation was observed in the presence of 5 mM Zn2+, Cd2+, and Pb2+. On the other hand, activity was enhanced by Ca2+ and Mn2+ at a concentration of 1 and 5 mM. Ca2+ was especially effective as an activator, increasing the activity more than sevenfold at a concentration of 5 mM. Significant inhibition was observed in the presence of the divalent metal chelator EDTA. The effects of detergents on the activity of LipTK-3 were also examined (Table 2). The tested detergents did not inhibit the activity of LipTK-3 at a concentration of 0.005 %. In fact, enzymatic activity was increased by 113 % using SDS, by 19 % using Tween 20, and by 11 % using Triton X-100. The effects of various organic solvents on the stability of LipTK-3 was examined (Table 3). After treatment in 20 % organic solvent for 1 h, the lipolytic activity of the enzyme demonstrated relatively stable activities in ethanol, isopropanol, dimethylsulfoxide (DMSO), and methanol.
Discussion
A psychrotrophic Pseudomonas sp. TK-3 was isolated from dirty and cool stream water. We cloned and characterized a cold-adapted lipase from the bacterial strain which was shown to belong to subfamily Ι.3. Pseudomonas sp. lipase LipTK-3 exhibited optimal activity at 20–25 °C towards p-nitrophenyl caprylate at the optimal pH of 8.0, had about 30 % of its maximal activity at 5 °C, but it was heat-unstable above 25 °C, indicating that the enzyme bears the typical characteristic of a cold-adapted enzyme [21]. Lipase from strain TK-3 showed high amino acid sequence homology to LipB52 from P. fluorescens B52 [22], LipB68 from P. fluorescens B68 [11], KB-Lip from Pseudomonas sp. KB700A [5], and TliA from P. fluorescens SIK W1 [23]. Interestingly, the optimal temperatures of these homologous lipases varied over a wide range; the optimum temperatures of LipB68, KB-Lip, LipB52, and TliA were reported to be 20 °C [11], 35 °C [5], 40 °C [24], and 45–55 °C [25], respectively. Therefore, the optimal temperature of LipTK-3 is lower than for most other homologous lipases. From these results, we speculate that the considerable difference in optimal temperature for lipase activity may arise from minor differences in the amino acid sequences between LipTK-3 and the other homologous lipases.
The primary structure of LipTK-3 indicated that it was a member of subfamily Ι.3 of bacterial lipases according to the classification and properties of bacterial lipases reported previously [12, 13]. The deduced amino acid sequence of LipTK-3 is very similar (78–96 %) to those of subfamily Ι.3 lipase. Furthermore, the N-terminal domain contained the catalytic triad (Ser207, Asp255, and His313) and the GXSXG motif, while the C-terminal domain contained five repeats of a nine-residue GGXGXDXUX sequence motif and putative secretion signals near the C terminus, as well as subfamily Ι.3 lipases from P. fluorescens B52 [22], P. fluorescens B68 [11], Pseudomonas sp. KB700A [5], and P. fluorescens SIK W1 [23]. Several repeats of the GGXGXDXUX sequence motif form a β-roll motif in the presence of Ca2+ [13]. Recently, it has been shown that the β-roll motif probably acts as an intramolecular chaperon that facilitates folding of the N-catalytic domain [26, 27].
LipTK-3 preferably hydrolyzed tricaprylin (C8:0) and p-nitrophenyl caprylate (C8) among the triglycerides and p-nitrophenyl esters, respectively. It is noteworthy that the enzyme activities towards triglycerides and p-nitrophenyl esters may be affected by the solubility of the substrates. Nonetheless, it seems obvious that LipTK-3 has a preference for the medium chain substrates rather than the short and long chain substrates. In contrast, KB-Lip from strain KB700A [5] showed high activity towards tricaprin (C10:0) among the triglycerides, whereas TliA from strain SIK W1 [25] preferred tricaproin (C6:0) and tricaprylin (C8:0). Among p-nitrophenyl esters, p-nitrophenyl caprate (C10) was the preferable substrate to KB-Lip from strain KB700A [5] and LipB68 from strain B68 [11]. These results imply that there are some structural and functional differences between LipTK-3 and the other homologous lipases. While the other homologous lipases have not been studied for the enzyme activities towards natural oils, LipTK-3 showed high activity towards natural oils including vegetable oils and animal fats. Incubation of LipTK-3 with various metals showed that Co2+, Mg2+, and Mn2+ did not inhibit the activity of LipTK-3. Complete inhibition was obtained with Cd2+, Pb2+, and Zn2+. The specific activity was markedly increased with the addition of Ca2+ over 1 mM, whereas it was significantly inhibited in the presence of over 1 mM of the divalent metal chelator EDTA. It has been reported that all lipases of subfamily Ι.3 are strongly inhibited by EDTA and their activities are highly dependent on Ca2+ [5, 10, 25]. This could be due to the C-terminal domain of family I.3 lipases, which contains the repetitive nine-residue sequences and needs to be folded in order for the whole protein to fold into its active conformation. Moreover, LipTK-3 was able to retain a high level of activity in the presence of detergents. Indeed, its activity was actually increased by Tween 20, Triton X-100, and SDS at a concentration of 0.005 %. These results are similar to those of KB-Lip from strain KB700A [5] and indicate that these cold-adapted lipolytic enzymes may be useful in detergents. Stability in the presence of organic solvents is a requisite property of enzymes used in non-aqueous systems [28]. Since LipTK-3 was relatively stable in some of the tested organic solvents, this lipase may be useful for organic synthesis. The effects of organic solvents on the stability of the other homologous lipases have not been reported.
In the present study, LipTK-3 exhibited high activity at low and moderate temperatures and was very active and stable in the presence of several detergents, metal ions, and organic solvents. These properties may offer potential economic benefits and are expected to be useful for some industrial applications, such as for wastewater treatment, bioremediation in fat-contaminated cold environments, and additives in detergents for cold washing.
References
Jaeger, K. E., Ransac, S., Dijkstra, B. W., Colson, C., van Heuvel, M., & Misset, O. (1994). FEMS Microbiology Review, 15, 29–63.
Jaeger, K. E., Dijkstra, B. W., & ReetZ, M. T. (1999). Annual Review of Microbiology, 53, 315–351.
Jaeger, K. E., & Eggert, T. (2002). Current Opinion in Biotechnology, 13, 390–397.
Hasan, F., Shah, A. A., & Hameed, A. (2006). Enzyme and Microbial Technology, 39, 235–251.
Rashid, N., Shimada, Y., Ezaki, S., Atomi, H., & Imanaka, T. (2001). Applied and Environmental Microbiology, 67, 4064–4069.
Choo, D. W., Kurihara, T., Suzuki, T., Soda, K., & Esaki, N. (1998). Applied and Environmental Microbiology, 64, 486–491.
Kulakova, L., Galkin, A., Kurihara, T., Yoshimura, T., & Esaki, N. (1999). Applied and Environmental Microbiology, 65, 611–617.
Zhang, J. W., & Zeng, R. Y. (2008). Marine Biotechnology, 10, 612–621.
Zheng, X., Chu, X., Zhang, W., Wu, N., & Fan, Y. (2011). Applied Microbiology and Biotechnology, 90, 971–980.
Amada, K., Haruki, M., Imanaka, T., Morikawa, M., & Kanaya, S. (2000). Biochimica et Biophysica Acta, 1478, 201–210.
Luo, Y., Zheng, Y., Jiang, Z., Ma, Y., & Wei, D. (2006). Applied Microbiology and Biotechnology, 73, 349–355.
Arpigny, J. L., & Jaeger, K. E. (1999). Biochemical Journal, 343, 177–183.
Angkawidjaja, C., & Kanaya, S. (2006). Cellular and Molecular Life Sciences, 63, 2804–2817.
Rosenau, F., & Jaeger, K. E. (2000). Biochimie, 82, 1023–1032.
Joseph, B., Ramteke, P. W., & Thomas, G. (2008). Biotechnology Advances, 26, 457–470.
Suzuki, T., Nakayama, T., Kurihara, T., Nishino, T., & Esaki, N. (2001). Journal of Bioscience and Bioengineering, 92, 144–148.
Laemmli, U. K. (1970). Nature, 227, 680–685.
Sommer, P., Bormann, C., & Götz, F. (1997). Applied and Environmental Microbiology, 63, 3553–3560.
Winkler, U. K., & Stuckmann, M. (1979). Journal of Bacteriology, 138, 663–670.
Lee, H. K., Ahn, M. J., Kwak, S. H., Song, W. H., & Jeong, B. C. (2003). Journal of Microbiology, 41, 22–27.
Feller, G., & Gerday, C. (2003). Nature Reviews Microbiology, 1, 200–208.
Tan, Y., & Miller, K. J. (1992). Applied and Environmental Microbiology, 58, 1402–1407.
Ahn, J. H., Pan, J. G., & Rhee, J. S. (1999). Journal of Bacteriology, 181, 1847–1852.
Jiang, Z., Zheng, Y., Luo, Y., Wang, G., Wang, H., Ma, Y., et al. (2005). Molecular Biotechnology, 31, 95–101.
Lee, Y. P., Chung, G. H., & Rhee, J. S. (1993). Biochimica et Biophysica Acta, 1169, 156–164.
Angkawidjaja, C., You, D. J., Matsumura, H., Kuwahara, K., Koga, Y., Takano, K., et al. (2007). FEBS Letters, 581, 5060–5064.
Meier, R., Drepper, T., Svensson, V., Jaeger, K. E., & Baumann, U. (2007). Journal of Biological Chemistry, 282, 31477–31483.
Ogino, H. (2009). Seikagaku, 81, 1109–1118.
Acknowledgements
We thank Dr. S. I. Aizawa of the Prefectural University of Hiroshima for assistance with transmission electron microscopy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tanaka, D., Yoneda, S., Yamashiro, Y. et al. Characterization of a New Cold-adapted Lipase from Pseudomonas sp. TK-3. Appl Biochem Biotechnol 168, 327–338 (2012). https://doi.org/10.1007/s12010-012-9776-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-012-9776-7