
Abstract
Two DNA fragments containing the entire coding sequences of lactate dehydrogenase (LDH; ldhL1 and ldhD), whose enzymes have high activity for bioconversion of phenylpyruvate (PPA) to phenyllactate (PLA), were amplified from Lactobacillus plantarum SK002 using PCR. Sequencing showed open reading frames of 963 bp (ldhL1) and 999 bp (ldhD) encoding putative proteins of 320 and 332 amino acid residues, respectively. The LDH genes were cloned into an expression vector pET-22b(+) and expressed in Escherichia coli BL21(DE3). The purified recombinant l1-LDH and d-LDH had approximate (SDS-PAGE) molecular weights of 35 and 40 kDa, respectively. l1-LDH and d-LDH had PPA bioconversion specific activities of 71.06 and 215.84 U/mg with K m values of 3.96 and 5.4 mM, respectively. The rl1-LDH and rd-LDH showed maximum enzyme activity at 30 and 40 °C while both had optimum activity at pH 6.0. l1-LDH exhibited a higher pH and temperature stability than d-LDH. The results show that the his-tagged L. plantarum SK002 d- and l1-LDHs are efficient catalysts for bioconversion of PPA to PLA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phenyllactate (2-hydroxy-3-phenyl-propionic acid or PLA) is a novel antimicrobial compound with broad inhibitory activity against a range of food spoilage fungal and bacterial species (both gram positive and gram negative) [1, 2]. Lactobacillus plantarum has extensively demonstrated PLA-based antimicrobial activity in over 10 strains [3–5].
Previously, PLA was mainly produced from bioconversion of phenylallanine (Phe) through amino acid metabolism [6]. It was however shown that phenylpyruvate (PPA) is potentially a more suitable substrate for PLA production [7, 8]. Recently, lactate dehydrogenase (LDH) was demonstrated to be the main enzyme for bioconversion of PPA to PLA in Lactobacillus spp. [9]. Different isoforms of LDH have been studied in Lactobacillus spp. as well as across strains in order to find one suitable for commercial use [10, 11]. The two LDH isoforms, l-LDH (EC 1.1.1.27) and d-LDH (EC 1.1.1.28) however have different evolutionary origins, catalytic properties, substrate specificities, and the presence of the l- and d-LDH isoforms in Lactobacillus spp. widely varies between strains with some strains having one or both of l- and d-LDH [12, 13].
According to the L. plantarum WCFS1 genome [14], there are three lactate dehydrogenases genes (ldhD, ldhL1 and ldhL2) which may occur mutually exclusively. L. plantarum SK002, with a high output of PLA from PPA was selected in our study. Recently, we demonstrated that the product of ldhL2 (L2-LDH) in L. plantarum SK002 has capacity for bioconversion of PPA to PLA but with low activity and specificity [15]. In this study, the other two LDH genes (ldhD, ldhL1) from L. plantarum SK002 were cloned, expressed in recombinant Escherichia coli BL21(DE3)/pET-ldhL1 and E. coli BL21(DE3)/pET-ldhD and then characteristics of their bioconversion of PPA to PLA were assessed.
Materials and Methods
Strains, Plasmids, Enzymes, and Chemicals
L. plantarum SK002, with a high output of PLA from PPA, was isolated and kept in our lab frozen at −70 °C. The E. coli strains JM109 and BL21(DE3) (Shanghai Sangong Biological Engineering Technology and Services Co. Ltd, Shanghai, China) were used as host strains for genetic cloning and expression, respectively. Restriction endonuclease, T4 DNA ligase, Taq DNA polymerase, and isopropyl-1-thiobeta-d-galactopyranoside (IPTG) were obtained from Takara Biotechnology Co. Ltd (Dalian, China). The plasmid pMD-T (Takara Biotechnology Co. Ltd Dalian, China) and pET-22b(+) (Novagen, USA) were used for cloning and expression of the lactate dehydrogenase genes in E. coli. Ni Sepharose Fast Flow (1.0 × 10.0 cm) was obtained from Amersham Biosciences (Piscataway, USA) and electrophoresis reagents were purchased from Bio-Rad (California, USA). All other chemicals were of reagent grade and were obtained from the local commercial sources.
Amplification by PCR and Subsequent Cloning of ldhD and ldhL1 Genes
The genomic DNA of L. plantarum SK002 was obtained using the genomic DNA purification kit from Sangong (Shanghai, China) then used as a template for the polymerase chain reaction (PCR). The oligonucleotides were designed according to the nucleotide sequence of the lactate dehydrogenase genes of L. plantarum WCFS1. The oligonucleotides for ldhL1 incorporating NdeІ and XhoI restriction sites were GCGCCCATATGTCAAGCATGCCAAATC (P1) and GCCGCCCTCGAGTTTATTT TCTAATTCAGC (P2), respectively. For ldhD, P1 and P2 incorporating EcoRI and XhoI respective restriction sites were CGGGGGAATTCATGAAAATTATTGCATAT and GCCGCTCGAGGTCAAACTTAACTTGCGTAT. The reaction mixtures for PCR contained 1× PCR buffer, each deoxynucleoside triphosphate (dNTP; 160 μM), each primer (0.4 μM), DNA template (1 ng/μl) and Taq DNA polymerase (1 U), all in a final volume of 20 μl. DNA amplification was started at 94 °C (5 min) and recycled 35 times as follows: 94 °C (60 s) and 60 °C (60 s) for ldhL1 or 54 °C for ldhD (60 s), 72 °C (90 s), and a final extension at 72 °C (10 min). Verification of the PCR products was done using electrophoresis in 1.0% agarose gel stained with ethidium bromide. The DNA fragments of interest (963 bp for ldhL1 and 999 bp for ldhD) were purified by the agarose gel electrophoresis and then ligated into pMD-T cloning vector. The recombinant DNA was used to transform E. coli JM109. The clones harboring recombinant plasmids pMD-ldhL1 and pMD-ldhD were screened and verified by gene sequencing. The plasmids were isolated using the plasmid isolation kit from Sangong (Shanghai, China). The ldhL1 gene fragment was digested with NdeI and XhoI from pMD-ldhL1. The ldhD gene fragment, however, was digested with EcoRI and XhoI from pMD-ldhD. The ldhL1 or ldhD gene fragments were subcloned into the plasmid pET-22b(+) between NdeI or EcoRI sites and XhoI for both, to yield pET-ldhL1 or pET-ldhD, respectively. The LDH genes were expressed under control of T7 lac promoter. The recombinant E. coli BL21(DE3)/pET-ldhL1 or E. coli BL21(DE3)/pET-ldhD were finally constructed by transforming the pET-ldh into the host strain BL21(DE3) using the CaCl2 method. The correct cloning of the LDH genes was confirmed in all cases both by restriction enzyme analysis and sequencing.
Expression and Purification of Recombinant d-LDH and l1-LDH
For expression of the recombinant LDHs, the transformed E. coli BL21(DE3) cells were grown in 400 mL of LB medium containing ampicillin (50 μg/ml) at 37 °C and 200 rpm. Induction was done at OD600 = 0.3 with 0.4 mM of IPTG. Then the cells further grown for an additional 12 h at 16 °C 180 rpm to OD600 = 1.0 to express active enzymes, and then harvested by centrifugation at 4 °C for 10 min at 10,000 rpm.
To purify the recombinant LDHs, the cell pellets were resuspended in 20 mM phosphate buffer (pH 7.4) to 0.1 of the original broth volume, disrupted by sonication at 4 °C (600 W, pulse on, 1 s; pulse off, 2 s) for 10 min and then the cell debris were removed by centrifugation (15,000 rpm, for 30 min at 4 °C). The cell free extract was applied onto a Ni Sepharose Fast Flow previously equilibrated with binding buffer (20 mM phosphate buffer, 500 mM NaCl, pH 7.4) [16]. Unbound proteins were washed out with washing buffer (20 mM phosphate buffer, 500 mM NaCl, 100 mM imidazole, pH 7.4). The recombinant LDHs were eluted from the column with elution buffer (20 mM phosphate buffer, 500 mM NaCl, 500 mM imidazole, pH 7.4). The eluted fractions were dialyzed overnight against 20 mM phosphate buffer (pH 7.4) at 4 °C then assessed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (12%). The purified LDHs were stored at 4 °C before usage.
Enzymatic Characteristics of Purified Recombinant d-LDH and l1-LDH
The activity of the LDHs towards PPA and pyruvate was routinely determined by measuring the rate of disappearance of NADH at 340 nm [9, 17]. The assay mixture contained in 100 mM potassium phosphate buffer (pH 6.5) 0.6 μmol of NADH, 19.6 μmol of PPA or 2.27 μmol of pyruvate and relevant amounts of enzyme (among the range of 50–300 U/l) in a total volume of 3.0 ml. The enzyme assay was performed at 30 °C, and 1 U of enzyme activity was defined as the amount of enzyme that catalyzes the degradation of 1 μmol of NADH per minute [9]. The effect of pH on the PPA to PLA bioconverting activity of the rLDHs was measured in 100 mM of citrate phosphate buffer (pH 2.0), sodium acetate buffer (pH 3.0–5.0), potassium phosphate buffer (pH 6.0–8.0), Tris–HCl buffer (8.0–9.0), and glycine–NaOH buffer (pH 10.0). The pH stability of the LDHs was tested by pre-incubating the enzyme at 30 °C for 60 min in different buffers at the indicated pH values. For determining the optimum temperature, enzyme activity was measured at different temperatures ranging from 20 to 70 °C. To examine the thermal stability, the enzyme was pre-incubated at temperatures ranging from 20 to 70 °C for different periods of time before assay at 30 °C as described previously. To determine the apparent Michaelis–Menten constant (K m), enzyme activities using PPA and pyruvate were measured by varying the concentrations of each substrate under optimum conditions of pH and temperature. The K m values of the LDHs were calculated by non-linear curve fitting using OriginLab software (Massachusetts, USA) [18, 19].
Reverse Phase HPLC Analysis of PLA and PPA
The product obtained from PPA reduction by purified recombinant l1-LDH and d-LDH was confirmed using reverse phase high-performance liquid chromatography (HPLC) equipped with an Agilent Zorbax XDB-C18 column (4.6 × 150 mm, 5 μm) using diode array detector (Agilent 1200 series) at 210 nm [9]. Briefly, for enzymatic production of PLA, 2 mM PPA, 1 mM NADH, and about 2,000 U/l purified enzyme were dissolved in 100 mM potassium phosphate buffer (pH 6.5) in a total volume of 1 ml. The reaction mixture was maintained at 30 °C for 1 h and then terminated by the addition of 1 M HCl. The reaction mixture (10 μl) was then loaded to HPLC. Elution was performed with methanol/0.05% TFA (solvent A) and water/0.05% TFA (solvent B) at 1 ml/min and A/B ratios of 10:90, 100:0, 100:0, and 10:90, with run times of 0, 20, 23, and 25 min, respectively.
Results and Discussion
Cloning and Sequence Alignment
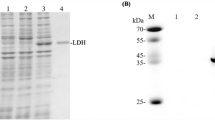
The oligosaccharide primers for PCR were designed from the LDH genes (ldhL1 and ldhD) from L. plantarum WCFS1. Using the genomic DNA of L. plantarum SK002 as the template, specific DNA fragments matching the sizes of ldhL1 and ldhD were amplified (Fig. 1). The fragments were ligated into the pMD-T cloning vector, and the recombinant plasmids pMD-ldhL1 and pMD-ldhD sequenced. Open reading frames of 963 bp (ldhL1) and 999 bp (ldhD) encoding putative proteins of 320 and 332 amino acid residues were found, respectively.

PCR amplification of the ldhD and ldhL genes. Lane 1 DNA standards, lane 2 and 3 ldhD, lane 4 nd 5 ldhL l
The genebank accession numbers for the genes were FJ712707 (ldhD) and FJ712706 (ldhL1). Based on putative amino acids, l1-LDH and d-LDH had a homology of 18%. The putative d-LDH and l1-LDH had a homology of 14.95% and 54% with l2-LDH [15], respectively. This indicates that L. plantarum SK002 contains three distinct LDH isoenzymes. It confirms the presence of two ldhL genes in L. plantarum in addition to the ldhD gene. Given the low homology of ldhL1 and ldhL2 with ldhD, the two are probably of different evolutionary origin from the ldhD gene as suggested by Taguchi et al. [20]. The two ldhL genes were as a result of duplicating events as recently explained by Cristescu et al. [21].
Sequence comparison of ldhL1 and ldhD to those from L. plantarum WCFS1 respectively showed 99% similarities at DNA level for both. Comparison of the amino acid sequence encoded by ldhD to that of other ldhD genes whose enzymes convert PPA to PLA showed that the highest identity was with L. plantarum ATCC 8041 (95%, ac. BAA14352), Lactobacillus delbruekii (53.92%, ac. YP_812208), Lactobacillus fermentum (53.44%, ac. YP_001843448), Lactobacillus acidophilus (52.94%, ac. YP_192990), and Lactobacillus brevis (32.59%, ac.YP_794398). The ldhL1 gene on the other hand showed an identity similarity of 76.21% (ac. YP_794703), 63.88% (ac. YP_001576807), and 52.38% (ac. YP_193798) in amino acid sequence to l-LDH genes of L. brevis, Lactobacillus helveticus, and L. acidophilus, respectively.
Expression and Purification of the LDH Enzymes
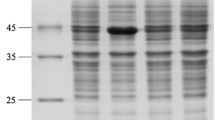
The two enzymes were overexpressed using the pET-vector system under the T7 lac promoter in E. coli BL21(DE3) cells (Fig. 2). The enzymes were purified by centrifugation, cell sonication, and chelating using a Ni Sepharose Fast Flow as described above (Fig. 3). The SDS-PAGE patterns of the his-tagged recombinant enzymes showed approximate molecular weights of 35 kDa (l1-LDH) and 40 kDa (d-LDH) which were similar to the predicted values based on the putative amino acid sequences.

Physical maps of expression plasmid pET-ldhD (a) and pET-ldhL1 (b) T7 lac T7 lac promoter, ori origin of replication, lacI the mutant repressor gene of lac operon, amplifier. The transcriptional direction of the LDH genes is indicated with arrows

SDS-PAGE analysis of the expression and purification of recombinant D and L1-LDH from E. coli BL 21 (DE3). Lane M standard protein molecular weight markers (kDa); lane 1 and 5 purified recombinant d-LDH and L1-LDH, respectively; lane 2 and 4 periplasm extract of induced transform ant harboring pET-ldhD and pET-ldhL1, respectively; lane 3 periplasm extract if induced transform ant harboring the empty pET-22b(+) vector
Properties of Purified LDHs Catalyzing the Conversion of PPA into PLA
The l1-LDH showed a specific activity of 71.06 U/mg and 549.35 U/mg, while d-LDH demonstrated specific activity values of 215.84 U/mg and 2,682.14 U/mg for PPA and pyruvate, respectively. The ability of the purified enzymes to convert PPA to PLA was confirmed using reverse phase HPLC (Fig. 4). The specific activity of the recombinant l1-LDH on PPA was over 1,000 times that of l2-LDH reported earlier in L. plantarum SK002 [15]. The increase in activity is probably due to the low homology (54%) between them. The relatively low activity can further explain why there are no previous reports on the purification or separation of the two l-LDH isoforms in L. plantarum. The PPA to PLA bioconverting activity of the purified recombinant l1- and d-LDH were moderately high compared to other studies which have reported values ranging from a maximum of 593.404 U/mg for L. helveticus l-LDH [22] to a minimum of 0.06 U/mg for l2-LDH in L. plantarum SK002 [15]. The present study confirms the presence of three PPA to PLA bioconverting LDH isoforms in a single strain. L. plantarum therefore presents a formidable strain for assessing the evolutionary development of PPA to PLA bioconverting ability in Lactobacillus spp.

RP-HPLC analysis of PPA bioconversation to PLA by purified recombinant d-LDH (a) and L1-LDH (b) enzymes, respectively
Few studies have assessed the pH dependence of the PPA to PLA bioconverting activity and stability of Lactobacillus spp. LDH. The variation of activity of the two purified enzymes with pH was assessed in the pH range of 3.0–8.0 with PPA as the substrate. Both enzymes showed optimum activity at pH 6.0 (Fig. 5a). A similar optimum was reported for LDH from Lactobacillus sp. SK007 [9]. The recombinant enzymes were both inactivated beyond pH 8.0. The pH-activity profile of the purified recombinant LDHs was in accordance that of the non-stereospecific in LDH studies in Lactobacillus sp. SK007 [9]. Purified recombinant l1-LDH however, showed a wider pH-activity range compared to both d-LDH and Lactobacillus sp. SK007 LDH [9]. It retained over 40% of its optimum activity at low pH (3.0) compared to d-LDH. The pH stability profile of purified recombinant LDHs is shown in Fig. 6c. d-LDH rapidly lost its activity beyond the common optimum (pH 6.0) while l1-LDH was more stable (>80% activity, pH 3.0–9.0) like the Lactobacillus sp. SK007 LDH studied by Li et al. [9]. The variable pH stability of the two purified recombinant LDHs with in the same strain L. plantarum SK002, present evolutionary implications (divergence based on environmental pH adaptation) and indicate that the l1-LDH has promise for industrial application in PLA production.

Variation of purified recombinant d-LDH and L1-LDH activity with pH and temperature. a Effects of pH on the activity of recombinant d-LDH and L1-LDH. The activity assay was conducted at 30 °C in buffers of pH 3.0–8.0. The maximum activity at pH 6.0 was defined as 100%. b Effects of temperature on activity of recombinant d-LDH and L1-LDH. The LDH activity was measured in potassium phosphate buffer (pH 6.5) at 20–70 °C. The maximum activity at 30 and 40 °C for purified recombinant d-LDH and L1-LDH was defined as 100%, respectively

Stability of purified recombinant d-LDH and L1-LDH Thermo-stability of the recombinant d-LDH (a) and L1-LDH (b). The enzyme was pre-incubated at 20–70 °C in potassium phosphate buffer (pH 6.5). An aliquot was drawn every 60 min then subjected to the activity assay at pH 6 (30 °C). The activity of untreated recombinant d-LDH and L1-LDH was defined as 100%. c Effect of pH on stability of recombinant d-LDH and L1-LDH. LDH activity was measured at 30 °C after incubation at 30 °C for 60 min in buffer of pH 2.0–10.0. The activity of untreated recombinant d-LDH and L1-LDH was defined as 100%
The temperature-activity profiles were assessed from 20 to 70 °C with PPA as the substrate (Fig. 5b). The optimum temperature for l1-LDH and d-LDH was 40 °C and 30 °C, respectively. Based on bioconversion of PPA to PLA, the optimum temperature in other studies has been reported as 45 °C for d-LDH in Lactobacilus confusus 20196 [17] and 40 °C for the non-stereospecific LDH in Lactobacilus sp. SK007 [9], thereby indicating a general variation around 40 °C. l1-LDH retained over 70% of maximum activity when held at 60 °C for 4 h (Fig. 6b). d-LDH however completely lost activity after incubation at 40 °C for 2 h (Fig. 6a). The purified l1-LDH was more stable than LDH for bioconversion of PPA to PLA reported by Li et al. [9]. The higher stability recombinant l1-LDH from the L. plantarum SK002 makes it a better candidate for industrial application.
The kinetic properties of the recombinant LDHs enantiomers were assessed using PPA and pyruvate as substrates. Both enzymes had a higher specificity for pyruvate than PPA with l1-LDH showing an apparent K m value of 0.23 and 3.96 mM while d-LDH showed values of 0.06 and 5.4 mM for pyruvate and PPA, respectively. This was in agreement with previous studies (Table 1). Therefore for bioconversion of PPA to PLA, an appropriate LDH with a high PPA specificity and specific activity is required. The l1-LDH and d-LDH in this study are relatively good candidates. Notably, the L. plantarum SK002 d-LDH K m values for pyruvate and PPA were 20- and fourfold lower than those reported by Taguchi et al. for L. plantarum ATCC 8041 [20].
In conclusion, there are two other enantioselective lactate dehydrogenase genes (ldhD and ldhL1) in L. plantarum SK002. The two genes have a homology of 18% based on the putative amino acid sequence. The two genes were overexpressed in recombinant E. coli BL21(DE3)/pET-ldhD and E. coli BL21(DE3)/pET-ldhL1, respectively. The specific activities of recombinant his-tagged l1-LDH and d-LDH for bioconversion of PPA to PLA were 71.06 U/mg and 215.84 U/mg, respectively. The characterization of enzymatic activities showed that the L. plantarum SK002 d- and l1-LDH exhibited same optimum pH for activity but varied significantly in specificity, temperature optima, temperature stability, and pH stability. These results show that the his-tagged L. plantarum SK002 d- and l1-LDHs are efficient catalysts for bioconversion of PPA to PLA hence good future candidates for genetic manipulation to improve industrial bioconversion of PPA to PLA.
References
Ohhira, I. Kuwaki, S. Morita, H. Suzuki, T. Tomita, S. Hisamatsu, S. et al. (2004). Biocontrol Science, 9(3), 77–81.
Lavermicocca, P. Valerio, F. & Visconti, A. (2003). Applied and Environmental Microbiology, 69(1), 634–640. doi:10.1128/AEM.69.1.634-640.2003.
Valerio, F. Lavermicocca, P. Pascale, M. & Visconti, A. (2004). FEMS Microbiology Letters, 233(2), 289–295. doi:10.1111/j.1574-6968.2004.tb09494.x.
Strom, K. Sjogren, J. Broberg, A. & Schnurer, J. (2002). Applied and Environmental Microbiology, 68(9), 4322–4327. doi:10.1128/AEM.68.9.4322-4327.2002.
Coloretti, F. Carri, S. Armaforte, E. Chiavari, C. Grazia, L. & Zambonelli, C. (2007). FEMS Microbiology Letters, 271(2), 245–250. doi:10.1111/j.1574-6968.2007.00723.x.
Vermeulen, N. Ganzle, M. G. & Vogel, R. F. (2006). J. Agric. Food Chem, 54(11), 3832–3839. doi:10.1021/jf052733e.
Li, X. Jiang, B. & Pan, B. (2007). Biotechnology Letters, 29(4), 593–597. doi:10.1007/s10529-006-9275-4.
Mu, W. Chen, C. Li, X. Zhang, T. & Jiang, B. (2009). Bioresource Technology, 100(3), 1366–1370. doi:10.1016/j.biortech.2008.08.010.
Li, X. Jiang, B. Pan, B. Mu, W. & Zhang, T. (2008). Journal of Agricultural and Food Chemistry, 56(7), 2392–2399. doi:10.1021/jf0731503.
Gaspar, P. Neves, A. R. Shearman, C. A. Gasson, M. J. Baptista, A. M. et al. (2006). FEBS Journal, 274(22), 5924–5936. doi:10.1111/j.1742-4658.2007.06115.x.
Okano, K. Zhang, Q. Shinkawa, S. Yoshida, S. Tanaka, T. Fukuda, H. et al. (2009). Applied and Environmental Microbiology, 75(20), 462–467. doi:10.1128/AEM.01514-08.
Hummel, W. (1999). Trends in Biotechnology, 17(12), 487–492. doi:10.1016/S0167-7799(98)01207-4.
Razeto, A. Kochhar, S. Hottinger, H. Dauter, M. Wilson, K. S. & Lamzin, V. S. (2002). Journal of Molecular Biology, 318(1), 109–1. doi:10.1016/S00222836(02)00086-4.
Kleerebezem, M. Boekhorst, J. Kranenburg, R. Molenaar, D. Kuipers, O. P. Leer, R. et al. (2003). Proceedings of the National Academy of Sciences, 100(4), 1990–1995. doi:10.1073/pnas.0337704100.
Jia, J. Mu, W. Zhang, T. & Jiang, B. (2009). Food and Fermentation Industries, 35(5), 22–25.
Sambrook, J. & Russell David, D. W. (2002). Molecular Cloning: A Labaratory Manual, 3, 1252–1255.
Hummel, W. Schütte, H. & Kula, M. R. (1983). Applied Microbiology and Biotechnology, 18(2), 75–85. doi:10.1007/BF00500828.
Copeland, R. A. (2000). Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis (pp. 124–128). Wiley: New York.
Origin Lab Corporation. (2003). Origin Pro 7.5 SRI. Version 7.5776(B776). Northampton MA. 01060. USA. www.originlab.com
Taguchi, H. & Ohta, T. (1991). Journal of Biological Chemistry, 266(19), 12588–12594.
Cristescu, M. E. Innes, D. J. Stillman, J. H. & Crease, T. J. (2008). BMC Evolutionary Biology, 8(1), 268. doi:10.1186/1471-2148-8-268.
Savijoki, K. & Palva, A. (1997). Applied and Environmental Microbiology, 63(7), 2850–2856.
Tokuda, C. Ishikura, Y. Shigematsu, M. Mutoh, H. Tsuzuki, S. Nakahira, Y. et al. (2003). Journal of Bacteriology, 185(16), 5023–5026. doi:10.1128/JB.185.16.5023-5026.2003.
Arai, K. Kamata, T. Uchikoba, H. Fushinobu, S. Matsuzawa, H. & Taguchi, H. (2001). Journal of Bacteriology, 183, 397–400. doi:10.1128/JB.183.1.397-400.2001.
Acknowledgement
This work was supported by the grants from the National High Technology “863” Program of China, project No. 2006AA10Z334, the Natural Science Foundation of Jiangsu Province, project No. BK2008099 and BK2008003, the Research Program of State Key Laboratory of Food Science and Technology, Jiangnan University, project No. SKLF-MB-200804 and SKLF-TS-200805, and the Program for Innovative Research Team of Jiangnan University, project No. 2008CXTD01.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jia, J., Mu, W., Zhang, T. et al. Bioconversion of Phenylpyruvate to Phenyllactate: Gene Cloning, Expression, and Enzymatic Characterization of d- and l1-Lactate Dehydrogenases from Lactobacillus plantarum SK002. Appl Biochem Biotechnol 162, 242–251 (2010). https://doi.org/10.1007/s12010-009-8767-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-009-8767-9