
Abstract
A lectin-like protein from the seeds of Acacia farnesiana was isolated from the albumin fraction, characterized, and sequenced by tandem mass spectrometry. The albumin fraction was extracted with 0.5 M NaCl, and the lectin-like protein of A. farnesiana (AFAL) was purified by ion-exchange chromatography (Mono-Q) followed by chromatofocusing. AFAL agglutinated rabbit erythrocytes and did not agglutinate human ABO erythrocytes either native or treated with proteolytic enzymes. In sodium dodecyl sulfate gel electrophoresis under reducing and nonreducing conditions, AFAL separated into two bands with a subunit molecular mass of 35 and 50 kDa. The homogeneity of purified protein was confirmed by chromatofocusing with a pI = 4.0 ± 0.5. Molecular exclusion chromatography confirmed time-dependent oligomerization in AFAL, in accordance with mass spectrometry analysis, which confers an alteration in AFAL affinity for chitin. The protein sequence was obtained by a liquid chromatography quadrupole time-of-flight experiment and showed that AFAL has 68% and 63% sequence similarity with lectins of Phaseolus vulgaris and Dolichos biflorus, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lectins are defined as carbohydrate-binding proteins or glycoproteins that are able to agglutinate cells or to precipitate carbohydrates, without having enzymatic activity toward their carbohydrate [1–3]. Plant lectins have been well studied as a model of carbohydrate recognition. A number of different physiological roles have been proposed for these molecules: plant defense, symbiosis of rhizobia, and protein folding [4–6].
Legume lectins represent the largest and most thoroughly studied family of these proteins [7] and show a remarkable conservation in primary, secondary, and tertiary structures. Indeed, approximately 85% of each legume lectin sequence is devoted to structurally conserved regions [8]. Comparisons of these sequences and structures have established that differences in carbohydrate specificity appear to be primarily due to differences in amino acid residues residing in adjacent loops to the carbohydrate-binding site [9–12]. The conformation of these loops is determined by the presence of calcium and transition metal ions in the protein structure [13–15].
Acacia farnesiana is a member of the Leguminosae family, and it is included in the Mimosoideae subfamily. Legume lectins are a large family of homologous proteins that show a strong similarity with regard to amino acid sequence and tertiary structure; however, their carbohydrate specificities and quaternary structures vary widely [7]. In addition, Vinorama isolectins (VL2–VL4) have been purified from seeds of Acacia constricta (vinorama), and an immunoassay direct analysis of VL peptides showed homologous sequences to those reported in different lectins of the genus Phaseolus [3]. Although, the Phaseolus vulgaris (PHA) isolectins usually bind to 1,6-branched GlcNac containing N-glycans; a PHA-like lectin that binds to chitin can represent a more potent defense protein against insects, fungi, nematodes, and bacteria than the similar ones [7]. Chitin is a natural homopolymer composed of β(1–4)-linked N-acetylglucosamine (GlcNAc)n and is a major component of the exoskeleton of fungi (comprising up to 30% of fungi cell wall) and invertebrates (gastrointestinal cover) [16].
In the Mimosoideae subfamily, only the seed lectins from A. constricta (VL)[3], Parkia speciosa (PSL) [17], P. javanica [18], P. platycephala (PPL-1) [19], P. discolor (PDL) [20], and the chimerolectin from P. plathycephala (PPL-2) [21] have been isolated and characterized in some detail. Moreover, crystal structures are available only for the native PPL-1 (1ZGR) [22] and in complex with 5-bromo-4-chloro-3-indolyl-α-d-mannose (1ZGS) [23], and PPL-2 (2GSJ) [24].
The variations in quaternary structures of leguminous lectins stimulated some studies to investigate the oligomerization of lectins by different trials, as ultracentrifugation [25] and X-ray crystallography [26]. The Cratylia floribunda lectin (bCFL) was found to have a pH-dependent tetrameric structure. In bCFL, Asn131 makes intradimer contacts with residues Asn122 and Ala123. In comparison, a histidine residue at position 131 in the Dioclea grandiflora lectin establishes a network of interdimer interactions bridging the central loops from the four monomers, thereby stabilizing the pH-independent tetrameric structure [26].
Oligomerization states may also be observed by molecular exclusion chromatography as a simple technique of investigation. Our studies characterize and sequence by tandem mass spectrometry a purified chitin-binding lectin-like protein from the albumin fraction of A. farnesiana seeds, a protein isolated in Mimosoidae species, verifying the time-dependent oligomerization process.
Materials and Methods
Material
A. farnesiana seeds were collected from plants located at Pacheco beach, Ceará, Brazil. Rabbit erythrocytes were obtained from the Universidade Federal do Ceará (UFC), and human blood was obtained from healthy donors from the Hematology Center of UFC. Reagents were purchased from Sigma™ and GE Healthcare™.
Soluble Protein Extraction Procedures
Mature seeds from A. farnesiana were ground into a fine powder using a coffee mill. The flour was defatted with n-hexane and air dried at room temperature. The powder obtained was extracted with 0.5 M NaCl in a proportion of 1:10 (w/v) for 3 h at room temperature with continuous stirring. The material was centrifuged (10,000 × g, 20 min at 4 °C). The pellet was discarded, while the supernatant was used for the assays presented here. The soluble fraction was dialyzed exhaustibly for 24 to 48 h at 25 °C against H2O. The dialyzed suspension was then centrifuged (10,000 × g, 20 min at 4 °C), and the pellet (albumin fraction) was frozen and lyophilized for later use. Protein concentration was determined by the method described by Bradford [27], and bovine serum albumin (BSA) was used as the standard.
Hemagglutinating activity
Hemagglutinating activity was determined in tubes by serial dilution. All ten tubes in the series received 100 μL of 0.1 M Tris–HCl buffer, pH 7.6, containing 0.15 M NaCl. In the first tube of the series, 100 μL of the supernatant obtained in the step described above was added. The second tube received 100 μL from the mixture of the first and so on until the tenth tube accumulated 200 μL and was left out of the assay. Afterward, 100 μL of 2% rabbit erythrocyte suspension in the same buffer was added to each tube. Hemagglutination was determined after 1 h incubation at 37 °C. Hemagglutinating activity was expressed as a titer namely, the reciprocal of the highest dilution that gave a positive result. The specific hemagglutinating activity was expressed as a titer per milligram of protein.
Sugar Specificity
The lectin sugar specificity determination was tested by comparing the inhibitory activity of sugars and glycoproteins on hemagglutination. The initial concentration of carbohydrates and glycoproteins were 0.1 M and 10 mg/mL, respectively. The carbohydrates used included d-glucose, d-galactose, d-mannose, N-acetyl-d-glucosamine, N-acetyl-d-galactosamine, l-fucose, lactose, α-methyl-mannoside, chitooligosaccharides (two to eight GlcNAc residues), and ovomucoid. Results were expressed as the minimal concentration of sugar or glycoprotein required to inhibit hemagglutination.
Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis
Sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) was carried out in 2 mm vertical gel slabs of 12% polyacrylamide separation gel with 3 M Tris–HCl, pH 8.8, and 1% SDS buffer and 3.5% stacking gel with 0.5 M Tris–HCl, pH 6.8, and 1% SDS buffer. Samples were dissolved in 0.0625 M Tris–HCl, pH 8.3, and 1% SDS buffer with 5% 2-mercaptoethanol and 0.02% bromophenol blue and incubated at 100 °C for 10 min. Electrophoresis was conducted at a constant current of 20 mA for 4 h. The protein bands were visualized by staining with Coomassie Brilliant Blue R-250. The molecular markers were BSA (66.0 kDa), carbonic anhydrase (29.0 kDa), cytochrome c (12.4 kDa), and aprotinin (6.5 kDa).
Chromatography Procedures
Ion-exchange Chromatography
Ion-exchange chromatography was carried out in a column containing quaternary ammonium (Mono Q HR 5/5) equilibrated with 0.1 M Tris–HCl buffer, pH 7.0. The albumin fraction was applied directly to the anion-exchange column (1 mL). The column was washed with 0.1 M Tris–HCl buffer, pH 7.0, followed by elution with a linear gradient of NaCl (0–1 M) in 0.1 M Tris–HCl, pH 7.0. Protein detection was monitored at 280 nm, and 1-mL fractions were collected, dialyzed against ultrapure water (MilliQ, Millipore), and then freeze dried. Protein and hemagglutination titers were determined for each fraction.
Chromatofocusing
Chromatofocusing of the protein fraction obtained by ion-exchange chromatography was performed on a Mono P HR 5/5 column (Pharmacia Biotech), equilibrated with 0.025 M Tris–HCl buffer, pH 7.5. Elution was carried out at a flow rate of 1.0 mL/min with a linear pH gradient of 7 to 4 using Polybuffer and 0.25 M Tris–HCl buffer, pH 7. Protein detection was monitored at 280 nm, and fractions with 1 mL were collected, dialyzed against ultrapure water (MilliQ, Millipore), and then freeze dried.
Molecular exclusion and reverse-phase chromatography
Approximately 1 mg of the purified protein obtained by ion-exchange chromatography was dissolved in 200 μL 0.1 M sodium phosphate buffer, pH 7.4, containing 0.01 mM CaCl2. This step was followed by centrifugation at 4,500 × g for 5 min.
Approximately 75 μL of supernatant was recovered and incubated at 37 °C. After this procedure, 15 μL of the protein was applied on the high-pressure liquid chromatography (HPLC) molecular exclusion column (Protein Pack 300SW-Glass, 0.8 × 30 cm) previously equilibrated with the same dissolving buffer. Three chromatography runs were carried out at different injection times of samples to determine oligomerization properties and to estimate molecular mass. The first sample was injected at time zero after centrifugation followed by injections at 90 and 180 min. The whole-protein fraction purified in the first chromatography run was subjected to a second chromatographic step using reverse-phase HPLC. The fraction was dissolved in 200 μL of 0.1% trifluoroacetic acid (TFA; buffer A), followed by clarification using high-speed centrifugation (4,500 × g for 3 min). The supernatant was then injected into a μ-Bondapack C18 reverse-phase HPLC column (0.3 × 30 cm). To elute the protein, a nonlinear gradient of buffer A to buffer B (66.6% acetonitrile in 0.1% TFA) was performed at a constant flow rate of 1.0 mL/min. The run was monitored at 214 nm, and the fraction obtained was then lyophilized.
Chitin Affinity Chromatography
A sample of 50 mg of the purified protein was dissolved in 10 mL of 0.5 M NaCl and run through a chitin affinity chromatography column previously equilibrated with 0.5 M NaCl. The sample was left in the column for 4 h to improve binding. After washing out the unbound material, the bound protein was eluted with 0.1 M glycine at pH 9.0, containing 0.15 M NaCl. The material eluted was collected manually in 2-mL fractions and monitored at 280 nm. The protein peaks were dialyzed against water and lyophilized.
Amino Acid Composition
Amino acid analysis of A. farnesiana lectin-like protein (AFAL) was carried out with a System Gold (Beckman) Amino Acid Analyzer after hydrolysis at 110 °C for 24 h with 6 N HCl and for 4 h with 4 N HCl, respectively, in sealed and evacuated ampoules. After hydrolysis, HCl was removed by evaporation, and the residue was analyzed in a Biochrom 20, Pharmacia-LKB. After hydrolysis, the vial was opened and HCl, and the phenol was evaporated away under reduced pressure in the presence of NaOH. The hydrolysate was washed with Milli-Q water and dried under reduced pressure in the presence of PO5. After being dried, the sample was redissolved in sodium citrate buffer, pH 2.2, filtered through a 0.45-μm membrane (Millipore), and injected into the analyzer, which was previously standardized according to the commercial patterns of the 20 natural amino acids.
N-terminal Sequencing
Two milligrams of the purified protein obtained through molecular exclusion and reverse-phase HPLC was dissolved in 200 μL 6 M guanidine chloride (Merck, Darmstadt, Germany) containing 0.4 M Tris–HCl and 2 mM ethylenediaminetetracetic acid (pH 8.15). Nitrogen was blown over the top of the protein solution for 15 min, which was then reduced with dithiothreitol (6 M, 200 μL) and carboxymethylated with 14C-iodoacetic acid and cold iodoacetic acid. Nitrogen was again blown over the surface of the solution, and the reaction tube was sealed. This solution was incubated in the dark at 37 °C for 1 h and desalted using a Sephadex G25 column (0.7 × 12 cm) with 1 M acetic acid buffer. The reduced and carboxymethylated fraction (RC-Lec) was lyophilized and stored at −80 °C. Approximately 2 mol of the RC-Lec was dissolved in 100 μL of acetonitrile solution (66% acetonitrile in 0.1% TFA), and N-terminal determination was conducted using a Procise f automatic sequencer (Applied Biosystem). The phenylthiohydantoin (PTH) amino acids were identified by comparing their retention times to that of the 20 PTH amino acid standards. Peptides containing 14C-CM-Cys were monitored by detecting the radioactivity label using a liquid scintillation counter (Beckman model L-250).
Mass Spectrometry Analysis and Protein Sequencing by Tandem Mass Spectrometry
Molecular mass of the native and the reduced and carbamidomethylated or pyridylethylated AFAL was determined by matrix-assisted laser desorption/ionization–time-of-flight (MALDI-ToF) mass spectrometry using a PE Biosystems Voyager DE-PRO instrument operating at 20 kV accelerating voltage in the linear mode. The sample matrix was sinapinic acid.
Protein sequencing by mass spectrometry was obtained by the digestion of AFAL with trypsin, chymotrypsin, and thermolysin. An amount of 1 mg of AFAL was dissolved in 250 μL of a 4-mg/mL ammonium bicarbonate buffer and mixed for 1 min. The protein solution was mixed with 25 mL of 2 mg/mL trypsin, incubated for 3 h at 37 °C, and stored at −20 °C. Chymotrypsin digestion was performed in the same buffer as trypsin except that the pH was maintained at 8.0 at 23 °C. Thermolysin digestion was performed at a substrate/enzyme ratio of 50:1 (by mass) in 0.1 M Tris–HCl (pH 7.6) containing 0.05 M NaCl and 0.05 M CaCl2 at 37°C for 4 h.
The digested protein sample was diluted 100× with Milli-Q water, injected into a nanoelectrospray ionization source, and analyzed in a Micromass™ quadrupole time-of-flight instrument with a resolution of 8,000 and an accuracy of 10 ppm. The spectral data were processed using the Biolynx 4.0 program. The peptides were sequenced by the ProteinLynx and MASCOT program. The secondary structure prediction was carried by Advanced Protein Secondary Structure Prediction Server [29].
Results and Discussion
The soluble protein extract of the A. farnesiana seeds as well as the different protein fractions of albumin, globulin, glutelin, and prolamin showed hemagglutinating activity against rabbit erythrocytes, treated and nontreated with proteolytic enzymes. This activity was not inhibited by any of the tested carbohydrate compounds, either the methylated or (GlcNAc)n derivatives. A. farnesiana extract activity differed from that of A. constricta lectin, which shows hemagglutinating activity with human blood of the ABO system [3]. None of the chitooligosaccharides tested showed any reactivity. The same applies to ovomucoid, a glycoprotein rich in terminal nonreducing GlcNAc residues. These findings indicate that the specificity of AFAL could be possibly directed toward the core pentasaccharide of N-glycans described to plant lectins. Glycan array screening experiments could demonstrate that this specificity of lectin is possibly directed toward high-Man N-glycans comprising the core pentasaccharide of N-glycans. However, at present, no conclusions can be drawn with respect to the affinity of AFAL because it is difficult to inhibit its hemagglutination activity. This is quite similar to plant lectin, mainly those with an extended carbohydrate-binding site for N-glycans such as RobpsCRA [30].
The protein present in the albumin fraction from A. farnesiana seeds, obtained by ion-exchange chromatography, had a bound peak that eluted with 42% NaCl and showed hemagglutinating activity after 12 h (Fig. 1a). Amino acid analysis (Table 1) demonstrated that these proteins have a high content of aspartic acid (13.94%) and glutamic acid (12.19%) residues, and chromatofocusing established that AFAL has an isoelectric point at pH 4.0 ± 0.5 (Fig. 2).

Ion-exchange chromatography of AFAL. a The retained fraction from affinity chromatography was load on a Mono-Q HR 5/5 column. Elution was carried out at a flow rate of 1.0 mL/min with a linear gradient from 0 to 1 M NaCl in 0.25 M Tris–HCl buffer, pH 7.5. Protein elution was monitored at 280 nm. Asterisk indicates peak with hemagglutinant activity. b SDS-PAGE of the purified protein from A. farnesiana. Lane 1: molecular mass markers (BSA 66 kDa, chicken egg ovalbumin 45 kDa, glyceraldehyde 3-phosphate dehydrogenase 36 kDa, carbonic anhydrase 29 kDa, bovine pancreas trypsinogen 24 kDa, soybean trypsin inhibitor 20 kDa). Lane 2: AFAL

Chromatofocusing of AFAL. The retained fraction from ion-exchange chromatography was loaded on a Mono-P HR 5/5 column. Elution was carried out at a flow rate of 1.0 mL/min with a linear gradient pH range 7–4 Pollybuffer and 0.25 M Tris–HCl buffer, pH 7.5. Protein elution was monitored at 280 nm. Asterisk indicates peak with isoelectric point 4.0 ± 0.5
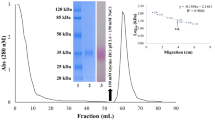
The purification of AFAL by ion-exchange chromatography was verified by SDS-PAGE, characterizing an electrophoretic profile consisting of two bands of apparent molecular mass of 50 and 29 kDa (Fig. 1b). The apparent molecular mass of AFAL determined by molecular exclusion chromatography was 120 kDa (Fig. 3), similar to that reported for the VL and PHA, which have an apparent molecular mass of 133 kDa [3, 28]. Mass spectrometry (MALDI-ToF) demonstrated a monomeric AFAL of 24.2 ± 1.0 kDa and a dimeric form of 50.0 kDa (Fig. 4), which indicates that AFAL is a tetrameric protein composed of identical subunits with the same apparent molecular mass (by chromatography and SDS-PAGE) and the same actual mass (by mass spectrometry).

Graph of molecular exclusion in which was used the protein in time zero (a), followed by a chromatography in column of reverse phase HPLC in μ-Bondapack C18 (0.3 × 30 cm; b. c, d SDS-PAGE of the purified protein deriving from albumin of A. farnesiana seeds peak I and peak II (c) of molecular exclusion chromatography and peak I (AFAL-RP) (d) of chromatography of reverse-phase HPLC in μ-Bondapack C18 (0.3 × 30 cm). Lane A: molecular mass markers (BSA 66 kDa, chicken egg ovalbumin 45 kDa, glyceraldehyde 3-phosphate dehydrogenase 36 kDa, carbonic anhydrase 29 kDa, bovine pancreas trypsinogen 24 kDa, soybean trypsin inhibitor 20 kDa). Lane B: AFAL

MALDI-ToF/mass spectra of the protein deriving from the fraction of A. farnesiana albumins (retained peak of ionic exchange). Asterisk, apparent molecular mass of 12.7 (double-charged ion), 24.2 (monomer), and 50.0 kDa (dimer)
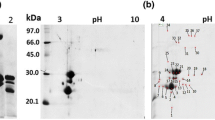
This new protein showed an N-terminal amino acid sequence very similar to that of A. constricta lectin and other lectins isolated from Phaseolus and Dolichos, but this similarity is much less when compared to Robinia lectin. This suggests that AFAL may be classified as a PHA agglutinin-like lectin. AFAL has an N-terminal with 87.5% similarity with the A. constricta lectin, a species that belongs to the Mimosoideae subfamily, and it does not show any similarity with regard to the N-terminal sequence of Parkia platycephala lectins (PPL and PPL2), also a Mimosioidae leguminosae (Fig. 5). However, AFAL displays a 62.1% similarity with the N-terminal portion of the PHA lectin (Phaseoleae subfamily). It has been postulated that the isolectins from PHA belong to a group of proteins with a conserved sequence and that part of the leguminous lectins have an evolutionary origin with common gene ancestors, probably to ensure and maintain important physiological functions [31]. This would indicate a link among lectins between the subfamilies of Mimosoideae and Papilionoideae.

N-terminal amino acid alignment of the A. farnesiana. These results showed an amino acid identity with other plant lectins of: 87.5%, 62.1%, 61.1%, 61.9%, 59.1%, 58.2%, 40.3%, and 40.3% with A. constricta lectin; PHAE-PHAU (P. vulgaris); PHA-L (P. vulgaris); LEC5_DOLBI (D. biflorus); LEC1_DOLBI (D. biflorus); PHAL_PHAUU (P. vulgaris); LCS2_ROBPS (Robinia pseudoacacia) and LCS1_ROBPS (R. pseudoacacia), respectively
The final AFAL sequence obtained by the overlap of sequenced peptides using tandem mass spectrometry showed 227 amino acid residues (Fig. 6), corresponding to a molecular mass of 25 kDa in accordance with the apparent molecular mass obtained with SDS-PAGE (29 kDa) and with the intact protein mass detected with MALDI-ToF (24 ± 1 kDa). The calculated pI for AFAL was 4.4, which is highly similar to the experimental pI of 4.0 ± 0.5. The alignment between AFAL and the National Center for Biotechnology Information databank revealed a similarity with some Phaseolae lectins. AFAL showed 68%, 66%, and 63% of similarity with PHA-leukoagglutinating isolectin (PHAL) [32], PHA hemagglutinin [33], and Dolichos biflorus lectin (DBL) [34], respectively. The amino acid analysis shown in Table 1 in comparison with the data obtained by mass spectrometry sequencing was very similar for all amino acid groups. The sequence alignment between AFAL, DBL, and PHA demonstrated at least five regions with a high degree of conservation (Fig. 7). The protein sequence data reported in this paper will appear in the UniProt Knowledgebase under the accession number P84849.

Amino acid sequence of AFAL obtained by tandem mass spectrometry. The peptides obtained and sequenced through enzymatic digestions are shown with letters (T) trypsin digestion, (Q) chymotrypsin, and (L) thermolysin in the overlaps

Sequence alignment among AFAL and the sequences of the PHA leucoagglutinating isolectin (PHAL), P. vulgaris hemagglutinin (PHAE), and D. biflorus lectin (DBL)
The high level of sequence conservation shows important conserved regions in AFAL sequence. The metal-binding sites remain conserved and are quite similar to PHA lectins with the amino acids consisting to four amino acid residues: Glu112, Asp114, Asp122, and His127 coordinating a Mn2+ ion and Asp114, Tyr117, Asn118, and Asp122 coordinating a Ca2+ ion. The carbohydrate-binding site also presents the same degree of conservation with the following amino acids: Phe06, Asn07, Glu08, Val19, and Glu185. A conserved hydrophobic sequence was observed in AFAL and PHA and PHAL (Val-Leu-Ile-Thr-Tyr). This sequence extends in the AFAL sequence from amino acid 147 to the 151.
A secondary structure prediction confirms the predominance of β-sheets motifs, 15 exactly, found in the legume lectins (see Fig. 7). The interfaces in legume lectins quaternary structures are built by conventional β-sheet-like contact between two strands, creating continuous, curved, antiparallel 15-strand β-sheet spanning two monomers, and the other interface is mainly formed by Van der Waals interactions between two β-strands [32].
Molecular exclusion chromatography showed that AFAL has two peaks with an apparent molecular mass of 40 and 80 kDa (Fig. 8), suggesting that this lectin undergoes oligomerization, which is supported by SDS-PAGE. It is possible to observe the same electrophoretic pattern in both peaks, demonstrating that it is the same protein. The molecular exclusion peak of 40 kDa was again shown with reverse-phase column chromatography. AFAL showed two peaks again with very close elution times and, therefore, with similar hydrophobicity, indicating that not only does this protein undergo oligomerization but it also has a relatively high quantity of exposed hydrophobic amino acids and is capable of binding to the column. The electrophoretic pattern of peak I in reverse-phase chromatography, which also shows two bands with apparent molecular mass of 29 and 59 kDa, is similar to the apparent molecular masses of the peaks that the AFAL displays with molecular exclusion.

Molecular exclusion chromatography of albumin fractions from A. farnesiana seeds in Protein Pack 300SW Waters Column. The oligomerization process is evidenced in AFAL at 37 °C in relation to the incubation time. AFAL immediately injected after diluted (a, Time 0). AFAL incubated in the buffer for 90 (b) and 180 min (c) at 37 °C
The chromatographic results showed that AFAL forms aggregated states, first in the dimeric form followed by a tetrameric form (Fig. 8). This aggregation may have great importance for this protein. Notably, some of the biological results were observed after 180 min or more of incubation (unpublished data). These results allow us to speculate that the aggregation of this protein may be important in enhancing biological activities by increasing its ability to bind polysaccharides. This protein appears to be a relatively hydrophobic compound, since it was eluted at a moderate acetonitrile concentration. The affinity pattern of AFAL with chitin is an interesting characteristic that can explain how protein oligomerization plays a role in carbohydrate-binding specificities of this protein.
Chitin affinity chromatography of the albumin fraction from A. farnesiana seeds showed an unbound peak (PI) and a bound peak (PII; Fig. 9a), where both peaks had hemagglutinating activity. SDS-PAGE of these peaks showed the same separation (Fig. 9b). Although, when PI and PII were submitted to molecular exclusion chromatography, they revealed the same chromatographic pattern (Fig. 9c). This pattern is characterized by two peaks with hemagglutinating activity. Therefore, the recognition and the interaction of the lectin molecules are specific for (GlcNAc)n polymers, and this recognition must occur through the extended carbohydrate-binding site, as described for the Curcubitaceae lectins [35–37].

Affinity properties of AFAL by chitin. a Chitin affinity chromatography of albumin fraction of A. farnesiana seeds show an unbound peak PIQ (dark gray shade) and a bound peak PIIQ (light gray shade). b Molecular exclusion chromatography of PIQ (dark gray shade) and the PIIQ (light gray shade) obtained in the affinity chromatography with the same pattern of elution. c SDS-PAGE. Lane a: molecular mass markers (BSA 66 kDa, chicken egg ovalbumin 45 kDa, glyceraldehyde 3-phosphate dehydrogenase 36 kDa, carbonic anhydrase 29 kDa, bovine pancreas trypsinogen 24 kDa, soybean trypsin inhibitor 20 kDa. Line b: peak I. Line c: peak II
AFAL has oligomerization properties, and this characteristic seems to modify its affinity binding properties. Molecular exclusion was carried out at different times of incubation of AFAL and the buffer. The chromatogram in Fig. 8 shows the elution at time 0, 90 and 180 min of incubation, and it is possible to see a gradual change in the main protein form, where AFAL is a monomer at time 0 and a dimer and tetramer with increasing incubation time. At time zero, two peaks represent a monomer with an apparent molecular mass of 40 kDa (large peak) and a dimer of 80 kDa (small peak; Fig. 8a). After 90 min, AFAL is predominantly in the dimer form (Fig. 8b) and shows three forms at 180 min of incubation with a high concentration of the tetrameric form (Fig. 8c).
These variations in the quaternary organization of AFAL enhance or reduce the affinity of this protein by chitin, indicating different biological actions for AFAL in different biological environments. The stability of quaternary structures were very well described for the lectins of Dioclea guianensis [38], D. grandiflora [25], and C. floribunda [24]. Following these studies, some residues of these lectins, which were positioned in the dimeric interface, are important to establish the structures by hydrogen bonds and Van der Waals interactions [38]. In a comparison between D. guianensis and D. grandiflora lectins, the latter lectin does not present dimer–tetramer equilibrium, which indicated that the substitution of His 131(Dgui) for Asn (DGL) drastically reduced interdimeric contacts and became unable to stabilize a specific loop (residues 117–123; [38]).
The agglutinin from PHA display other residues involved in interdimeric contacts. They are Ser186 and Lys184 from one monomer and Ser190 and Lys149 from another. In AFAL, based on the aligning between AFAL and PHA, these residues correspond to Tyr and Arg in one monomer and Ser and Arg from another (Fig. 10). The manipulation of these residues by site-directed mutagenesis and the tridimensional crystal structure could help to understand the time-dependent oligomerization process in AFAL.

Contacts between the dimers of PHA lectin. The amino acid residues Ser186 and Lys184 from one monomer (yellow) and Ser190 and Lys149 from another (blue) were involved in the dimeric interactions in PHA. In AFAL, these residues correspond to Tyr and Arg in monomer A and Ser and Arg in monomer B
Conclusion
AFAL is a chitin-binding protein purified from the albumin fraction of soluble extracts from A. farnesiana seeds. The complete amino acid sequencing obtained by tandem mass spectrometry confirms its similarity with PHA-like lectins. The time-dependent oligomerization of AFAL can explain the late hemagglutinating activity observed in our experiments and the variation in the affinity for chitin, which permits the elution of AFAL in both peaks of affinity chromatography. This is due to the time dependence of contact between the polysaccharide and AFAL. Once AFAL presents the recurrent variations in the quaternary structure assembly, this may enhance or minimize the chitin-binding properties leading to differential actions of AFAL in different cellular environments.
Abbreviations
- AFAL:
-
Acacia farnesiana lectin-like protein
- VL:
-
vinorama lectin
- GlcNac:
-
N-acetyl-glucosamine
- PPL-1:
-
Parkia platycephala lectin
- PPL-2:
-
Parkia platycephala chimerolectin
- bCFL:
-
Cratylia floribunda lectin
- BSA:
-
bovine serum albumin
- SDS-PAGE:
-
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- HPLC:
-
high-pressure liquid chromatography
- TFA:
-
trifluoroacetic acid
- EDTA:
-
ethylenediaminetetracetic acid
- DTT:
-
dithiothreitol
- PTH:
-
phenylthiohydantoin
- PHAL:
-
Phaseolus vulgaris leukocyte agglutinin
- DBL:
-
Dolichos biflorus lectin
- PDL:
-
Parkia discolor lectin
- PSL:
-
Parkia speciosa lectin
- PJL:
-
Parkia javanica lectin
References
Goldstein, I. J., Hughes, R. C., Monsigny, M., Osawa, T., & Sharon, N. (1980). Nature, 285, 66.
Van Damme, E. J. M., Peumans, W. J., Barre, A., & Rougé, P. (1998). Critical Reviews in Plant Sciences, 17, 575–692.
Guzman-Partida, A. M., Robles-Burgueno, M. R., Ortega-Nieblas, M., & Vazquez-Moreno, I. (2004). Biochimie, 86, 335–342.
Etzler, M. E. (1986). The lectins: Properties, functions and applications in biology and medicine. In I. E. Liener, N. Sharon, & I. J. Goldstein (Eds.) Distribution and function of plant lectins (pp. 371–435). New York: Academic.
Peumans, W. J., & Van Damme, E. J. (1995). Plant Physiology, 109, 347–352.
Helenius, A., Trombetta, E. S., Herbert, D. N., & Simons, J. F. (1997). Trends Cell Biology, 7, 193–200.
Sharon, N., & Lis, H. (2003). Lectins. Dordrecht, The Netherlands: Kluwer.
Imberty, A., Casset, F., Gegg, C. V., Etzler, M. E., & Perez, S. (1994). Glycoconjugate Journal, 11, 400–413.
Hamelryck, T. W., Moore, J. G., Chrispeels, M. J., Loris, R., & Wyns, L. (2000). Journal of Molecular Biology, 299, 875–883.
Loris, R., Hamelryck, T., Bouckaert, J., & Wyns, L. (1998). Biochimica et Biophysica Acta, 1383, 9–36.
Young, N. M., & Oomen, R. P. (1992). Journal of Molecular Biology, 228, 924–934.
Sharma, V., & Surolia, A. J. (1997). Molecular Biology, 267, 433–445.
Bouckaert, J., Loris, R., Poortmans, F., & Wyns, L. (1995). Genetics, 23, 510–540.
Bouckaert, J., Dewallef, Y., Poortmans, F., Wyns, L., & Loris, R. (2000). Journal of Biological Chemistry, 275, 19778–19787.
Lescar, J., Loris, R., Mitchell, E., Gautier, C., Chazalet, V., Cox, V., et al. (2002). Journal of Biological Chemistry, 277, 6608–6614.
Patil, R. S., Ghormade, V. V., & Deshpande, M. V. (2000). Enzyme Microbial Technology, 26, 473–483.
Suvachittanont, W., & Peutpaiboon, A. (1992). Phytochemistry, 319, 4065–4070.
Utarabhand, P., & Akkayanont, P. (1995). Phytochemistry, 38, 281–285.
Cavada, B. S., Santos, C. F., Grangeiro, T. B., Silva, L. I. M. M., Campos, M. J. O., Sousa, F. A. M., & Calvete, J. (1997). Physiology and Molecular Biology of Plants, 3, 109–115.
Cavada, B. S., Madeira, S. V. F., Calvete, J. J., Sousa, L. A. G., Bomfim, L. R., Dantas, A. R., et al. (2000). Preparative Biochemistry & Biotechnology, 30, 271–280.
Cavada, B. S., Castellón, R. E. R., Vasconcelos, G. G., Rocha, B. A. M., Bezerra, G. A., Debray, H., et al. (2005). Acta Crystallographica Section F, 61, 841–843.
Gallego Del Sol, F., Nagano, C., Cavada, B. S., & Calvete, J. J. (2005). Journal of Molecular Biology, 353, 574–583.
Gallego del Sol, F., Gomez, J., Hoos, S., Nagano, C. S., Cavada, B. S., England, P., et al. (2005). Acta Crystallographica Section F, 61, 326–331.
Cavada, B. S., Moreno, F. B., Rocha, B. A. M., Azevedo Jr., W. F. , Castellon, R. E., Goersch, G. V., et al. (2006). FEBS Journal, 273, 3962–3974.
Calvete, J. J., Thole, H. H., Raida, M., Urbanke, C., Romero, A., Grangeiro, T. B., et al. (1999). Biochimica et Biophysica Acta, 1430, 367–375.
Gallego Del Sol, F., Cavada, B. S., & Calvete, J. J. (2006). Journal of Structural Biology, 158, 1–9.
Bradford, M. M. A. (1976). Biochemistry, 72, 248–254.
Vázquez-Moreno, L., Ortega-Nieblas, M., Robles-Burgueño, M. R., & Ramos-Clamont, G. (2000). I.J.B.C, 5, 1–8.
Raghava, G. P. S. (2000). Comparative Asian Studies Programme, 4, 75–76.
Van Damme, E. J. M., Culerrier, R. I., Barre, A., Alvarez, R., Rougè, P., & Peumans, W. J. (2007). Plant Physiology, 144, 662–672.
Hemperly, J. J., & Cunningham, B. A. (1983). Trends in Biochemical Sciences, 5, 100–102.
Hamelryck, T. W., Dao-Thi, M. H., Poortmans, F., Chrispeels, M. J., Wyns, L., & Loris, R. J. (1996). Biological Chemistry, 271, 20479–20485.
Lioi, L., Sparvoli, F., Galasso, I., Lanave, C., & Bollini, R. (2003). Theoretical and Applied Genetics, 107, 814–822.
Hamelryck, T. W., Loris, R., Bouckaert, J., Dao-Thi, M. H., Strecker, G., Imberty, A., et al. (2000). Journal of Biological Chemistry, 286, 1161–1177.
Allen, H. J., & Johnson, A. Z. (1977). Carbohydrate Research, 58, 253–265.
Anantharam, V., Patanjali, S. R., Swamy, M. J., Sanadi, A. R., Goldstein, I. J., & Surolia, A. (1986). Journal of Biological Chemistry, 261, 14621–14627.
Peumans, W. J., Barre, A., Hao, Q., Rougé, P., & Van Damme, E. J. M. (2000). Trends in Glycoscience and Glycotechnology, 12, 83–101.
Wah, D. A., Romero, A., Gallego del Sol, F., Cavada, B. S., Ramos, M. V., Grangeiro, T. B., et al. (2001). Journal of Molecular Biology, 310, 885–894.
Acknowledgments
This work was partly financed by Fundação Cearense de Apoio ao Desenvolvimento Científico e Tecnológico (FUNCAP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Laboratório de Espectrometria de Massas/LNLS, Campinas—Brazil, and FAPESP (SMOLBNet, 01/07532-0). BSC and AHS are senior investigators of CNPq. We also thank Dr. A. Leyva for English language editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Santi-Gadelha, T., Rocha, B.A.M., Oliveira, C.C. et al. Purification of a PHA-Like Chitin-binding Protein from Acacia farnesiana Seeds: A Time-dependent Oligomerization Protein. Appl Biochem Biotechnol 150, 97–111 (2008). https://doi.org/10.1007/s12010-008-8144-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-008-8144-0