
Abstract
Lectin (TfgL) was purified from the seeds of Trigonella foenum-graecum (Fenugreek) belonging to fabaceae family by ammonium sulphate precipitation, ion exchange and followed by size exclusion chromatography. SDS–PAGE analysis revealed that TfgL molecular weight is approximately 27 kDa. 2D-PAGE reveals the existence of two isolectins (pI values of 6.3 and 6.7) with acidic nature and charge heterogeneity. The MALDI-TOF–MS and peptide mass fingerprinting investigation of TfgL showed sequence similarity with a lectin. The hemagglutinating activity of TfgL was stable in broad range of temperature 37–90 °C and at varied pHs 3, 7.6 and 10. Far-UV circular dichroism measurements showed that TfgL is mostly composed of α-helix (84.5%), β-sheet (6.5%), β-turns (5%) and unordered structures (4%). TfgL showed conformational stability in wide range of temperatures (20‒90 °C) and pHs (3, 7.6 and 10) but lost its secondary structure in the presence of 6 M Gdn.HCl. Quenching titrations were carried out with acrylamide and iodide quenchers in order to investigate the exposure and accessibility of the protein tryptophan residues. Maximum quenching observed with acrylamide compared to iodide revealed that the Trp residues of TfgL are buried in the protein core, which is hydrophobic in nature. TfgL showed binding affinity towards porphyrin, the association constant (Ka), for MnTSPP and MnTMPyP was calculated to be 1.2 × 106 M‒1 and 3.45 × 106 M‒1, respectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lectins are all over distributed in nature. They have been isolated from different kinds of species including viruses, bacteria, fungi, animals and plants (Mishra et al. 2019). In plants, the majority of lectins have been isolated from the seeds, where they accumulate in the process of maturation and vanish upon germination. In addition to seeds, lectins have also been found in nearly all types of tissue of plants. The plant lectins are very well known for binding properties towards carbohydrate and macromolecules (Lagarda-Diaz et al. 2017). Plant lectins have been classified into seven different families based on the sequence and structural motifs (Tsaneva and Damme 2020). Among them four families, namely the legume lectins, lectins containing hevein domains, monocot mannose binding lectins and type 2 ribosome inactivating proteins comprise abundant number of lectins, and are considered to be large families. Legume lectins showing sequence homology displays an unusual interspecies variability of sugar target selection. The members of lectin family proteins may consist of two or four subunits, either slightly identical or different, each with a single, small carbohydrate binding site with the same specificity (Sitohy et al. 2007).
Lectins can work as defensive components against the attack by fungus, bacteria, and insects as well as other predators (Silva et al. 2019). The hemagglutination activities are demonstrated by more or less all the lectins and are frequently found to be efficient antitumor, anti-HIV, natural immune modulators and also influence human lymphocytes proliferation (Mishra et al. 2019).
Porphyrins are used in photo dynamic therapy (PDT) as photosensitizers for the treatment of cancer (Kou et al. 2017), however, porphyrins selectivity towards tumor cells is normally insufficient to make sure their selective killing by the photodynamic action (Liang et al. 2020). In view of the identified capability that certain lectins can bind to tumor cells preferentially, it comes into view that lectins could work as specific targeting agents for porphyrin in PDT. Because of these important biological functions, in the recent years researchers have been paying attention on the purification and characterization of variety of lectins from a large number of sources.
Trigonella foenum-graecum (Fenugreek) is an annual herb belonging to legume family. The seeds are reported to have digestive stimulant action; control diabetic conditions and plasma cholesterol level, antioxidant properties and hepatoprotective effect (Srinivasan 2006).
The aim of present study is to purify and characterize lectin from Trigonella foenum-graecum (fenugreek) seeds, named as TfgL. The purification technique of TfgL is relatively simple, has the capacity to generate useful information quickly, safely, economically, and reproducibly. In this study TfgL hemagglutination activity and secondary structure stability towards temperature, pH and Gdn.HCl has been investigated. We have also examined the tryptophan exposure and environment in TfgL. The results revealed that the Trp residues are somewhat buried in native TfgL. We furthermore investigated porphyrin binding ability to TfgL for the application in PDT.
Materials and methods
Materials
Seeds of fenugreek were collected locally. Chromatographic and 2D-PAGE reagents were obtained from GE Healthcare. Molecular weight markers were purchased from Fermentas. Stirred Ultrafiltration Cell and 3 kDa cutoff membranes were obtained from Millipore, India. 5,10,15,20-Tetrakis(4-sulfonatophenyl)-21H,23H-porphine manganese(III) chloride (MnTSPP) and Manganese(III) 5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine chloride tetrakis (methochloride) (MnTMPyP) were purchased from Sigma-Aldrich. All other chemicals were purchased from Sigma-Aldrich of HPLC or analytical grade.
Isolation and purification of T. foenum-graecum lectin (TfgL)
30 g of T. foenum-graecum seeds free of seed coat were ground and removed fat by washing with hexane for 1 h, followed by four parts (w/v) of 1:2 ratio EtOH + CHCl3 for 30 min. This defatted flour was then extracted by using 1:10 (w/v) extraction buffer (25 mM sodium phosphate buffer, pH 6.5, 50 mM NaCl, 4 mM EDTA, 0.01% azide) at 4 ºC for 36 h. Homogenate was filtered by using muslin cloth then the filtrate was centrifuged at 12,000 rpm for 20 min at 4 ºC. The supernatant was named as crude extract (CE). This crude extract sample was first fractionated by ammonium sulfate precipitation, in which the crude extract was treated with (NH4)2SO4 to 60% saturation. Following centrifugation at 12,000 rpm at 4 ºC for 30 min, the supernatant was discarded while the pellet dissolved in 20 mM phosphate buffer, pH 7.6 was then dialyzed against the same buffer with numerous changes at 4 ºC in 3.5 kDa cutoff membrane (Spectra/Por6), then applied on XK16/20 column of DEAE-Sepharose using FPLC-AKTA Prime Plus system, earlier equilibrated with the equilibration buffer 20 mM phosphate buffer, pH 7.6. Subsequent to elution of unabsorbed proteins from column, absorbed proteins were eluted with linear gradient of 1 M NaCl in the identical buffer at a steady flow of 30 ml/hour. The absorbance of protein was observed at 280 nm. The unabsorbed fraction (D2) demonstrating hemagglutination activity was pooled, dialyzed against 20 mM sodium phosphate buffer, pH 7.6 at 4 °C and concentrated by Stirred Ultrafiltration Cell using the membrane with pore size of 3 kDa. This fraction was then applied to size exclusion chromatography on XK 16/100 column of Sephadex G-50 pre-equilibrated with 20 mM sodium phosphate buffer, pH 7.6. Fractions (3 ml) from the protein peaks were collected at a constant flow of 0.3 ml/min. The fractions (P4) showing hemagglutination activity were pooled and dialyzed against 20 mM phosphate buffer, pH 7.6 with several changes and further used for assays.
Protein quantification, SDS-PAGE and two-dimensional electrophoresis (2D-PAGE)
SDS–PAGE and native-PAGE (15%T, 4%C) was carried out by using the Laemmli method (Laemmli 1970). Visualization of protein bands were done by either 0.1% of Coomassie Brilliant Blue R-250 (CBBR) method or silver staining method (Blum et al. 1987). For the identification of pI value and number of isoforms, purified TfgL was first separated by isoelectric focusing in the Ettan IPGphor 3 system with 3–10 pH IPG strips (GE Healthcare) according to the method as previously described (Oddepally and Guruprasad 2015). The rehydration buffer consist of 2 M thiourea, 7 M urea, 1 M DTT, 4% (w/v) 3-[3-cholamidoproplyl (dimethylammonio)]- 1-propanesul- phonate (CHAPS), 1% bromophenol blue and 0.05% (v/v) IPG buffer 3–10 (GE Healthcare) and the IEF conditions were 300 V for 40 min, followed by gradient to 1000 V in 27 min, gradient to 5000 V in 80 min and 5000 V for 15 min at 20 ºC. The IPG strips were equilibrated for 15 min in a buffer solution containing 1 M Tris–HCl pH 8.8, 6 M urea, 30% (v/ v) glycerol and 2% (w/v) SDS. The second dimension was performed on polyacrylamide gels of 15% T and 4% C (Laemmli 1970).
Proteomic analysis: in-gel digestion and MALDI-TOF–MS analysis
In-gel digestion and matrix assisted laser desorption/ionization time of flight mass spectrometric (MALDI-TOF–MS) study was carried out with a MALDI-TOF/TOF mass spectrometer (Bruker Autoflex III smartbeam, Bruker Daltonics, Bremen, Germany) according to the process as described in our previous work (Oddepally et al. 2013). For data analysis, flex analysis software was utilized.
Protein classification: peptide mass fingerprinting analysis
Protein recognition was carried out by database searches, with MASCOT program (http://www.matrixscience.com) using Biotools software of Bruker Daltonics. The identification search for mass values was done by using existing digests values and sequence similarity from SwissProt and NCBInr database, and the category of taxonomy was set to Viridiplantae (green plants). The further search parameters were: fixed modification is carbamidomethyl (C), oxidation (M) as variable modification, trypsin as enzyme, peptide charge of + 1, monoisotropic and missed cleavage as -1. According to database searches by the MASCOT probability study (P < 0.05), only important hits were accepted for protein identification.
Hemagglutination assay
In hemagglutinating activity assay, a sequential two-fold dilution of the TfgL solution in microtiter U-plates (100 μl) was mixed with 4% suspension 100 μl of human RBCs in phosphate-buffered saline, pH 7.6 at 20 °C. The results were measured after about 1 h when the blank had completely sedimented. The hemagglutination titer is described as the minimum quantity of protein (mg/ml) that may stimulate visible erythrocyte agglutination and described as one hemagglutinating unit (HU). Specific activity was calculated as the number of hemagglutinating units per mg protein (Cavada et al. 2020).
The thermal stability of TfgL (1 mg/ml) was measured by incubation in the assay buffer (20 mM phosphate buffer, pH 7.6) at broad range of temperatures (37–100 °C) for 30 min. Subsequent to incubation, all the samples were kept on ice for 15 min prior to testing for remaining hemagglutination activity. Residual hemagglutination activity was analyzed in assay buffer, as described above. The pH effect on TfgL stability was estimated by studying the remaining activity after treatment at pHs 3, 7.6 and 10 at 25 °C for 30 min. Buffers used included Mcllvaine buffer (0.2 M Na-phosphate and 0.1 M Na-citrate) for pH 3.0 and 0.1 M glycine–NaOH for pH 10. All measurements were conducted in triplicate and yielded reproducible outcome.
CD spectroscopy
Circular dichroism measurements were carried out on a Jasco J- 810 spectropolarimeter, operational with a peltier-type temperature regulator at 25 ºC and a cell holder of thermostated, interfaced with a thermostatic tub. Far-UV (200–240 nm) CD spectra were measured in quartz cell of 1 cm path length at 0.8 mg/ml protein concentration in 50 mM sodium phosphate buffer, pH 7.0. Every CD spectrum was the average of three scans at 50 nm/min speed with 1 nm slit width, 1 s time response and 0.5 nm of data pitch. All samples were centrifuged prior to use and the clear supernatant was used for the studies. CD spectra were background corrected and scaled in molar ellipticity. The secondary structure investigation was carried out using CDNN software.
The effect of temperature on conformational stability of TfgL was investigated by recording CD spectra at temperatures in the wide range from 20 to 90 °C. The effect of pH on stability of TfgL was also evaluated by recording CD spectra at pHs 3, 7.6 and 10. Unfolding studies of TfgL were performed after treated with Gdn.HCl in different concentrations (0‒4 M) overnight prior to the experiment. Refolding studies of chemically denatured TfgL with 6 M Gdn.HCl were carried out by dilution with 20 mM phosphate buffer, pH 7.6 to different concentrations and incubated overnight prior to the experiment.
Fluorescence spectroscopy
All fluorescence measurements were done by Fluoromax-4 spectrofluorimeter. A slit width of 1 nm was usually used for the excitation and emission monochromators. The emission spectra of the protein solutions (in 10 mM phosphate buffer, pH 7.0) were measured in the region of 310‒440 nm at 25 °C showing an absorbance less than 0.1 at 280 nm after exciting at 295 nm. The temperature of the cell was maintained at 25 °C by thermostat. The sample reaction mixtures devoid of protein were utilized to the base line rectification. All samples were centrifuged before use and the clear supernatant was used for experiments.
Fluorescence intensity was measured with progressive addition of small aliquots of 5 M quencher stocks (acrylamide or potassium iodide) each time after 2 min period (allowing for equilibration). The final quencher concentration achieved in each case was 0.6 M. The potassium iodide stock solution had 0.2 mM sodium thiosulphate to avoid the formation of triiodide (I3−). Reaction mixtures without TfgL were used to correct the base line. Before using, all samples were centrifuged and the clear supernatant was used for the studies.
The quenching data was analysed by using Stern‒Volmer Eq. (1) as well as by using the modified Stern‒Volmer Eq. (2).
Where Fo and Fc are the respective fluorescence intensities, in the absence and presence of quencher, after corrected for dilution, correspondingly. [Q] is the quencher concentration, Ksv is the Stern‒Volmer constant for given quencher, fa refers to the part of the total fluorescence that is available to the quencher and Ka is the corresponding quenching constant.
Porphyrin binding studies
Porphyrin binding to TfgL was examined by the absorption titration method, which is described previously for Momordica charantia lectin, MCL (Sultan et al. 2004a, b). All titrations were carried out in 20 mM sodium phosphate buffer, pH 7.4. Porphyrin samples (2 µM) (A) MnTSPP, (B) MnTMPyP were titrated by addition of small aliquots of the TfgL. The same volume of the protein was added to the reference cell, to correct protein contribution to the absorbance. UV-Vis spectra were recorded after an equilibration interval of 2 min subsequent to each addition. The spectra were multiplied by a suitable factor to correct the dilution effect in the intensities resulting by the addition of protein. A plot of (A0/ΔA) vs (1/ [P]t), where A0 is the absorbance of the sample in the absence of protein and ΔA is the change in absorption intensity at the total protein concentration, [P]t, showed a straight line (Fig. 7C, D). From the ordinate intercept of this plot, A∞, the sample absorbance at infinite protein concentration was obtained. From this analysis, the association constant, Ka, characterizing the porphyrin–TfgL interaction is determined according to Eq. (3).
Where [P]f is the free protein concentration, is given by.
From the Eq. (3) the X-intercept of a plot of log [ΔA0 /(Ac ‒ A∞)] vs. log[P]f will yield pKa for the lectin– porphyrin association.
Statistical analysis
The quantification of protein yield during various purification stages, activity assays and their dependence on pH, temperature and salt concentration were carried out in triplicate. The statistical evaluation of the obtained data was undertaken by one-way Analysis of variance (ANOVA) followed by Bonferroni’s test for multiple comparisons by using Origin version 7 software. The probability (P = value) P ≤ 0.05 was considered statistically significant. The mean and standard deviations were reported.
Results and discussion
Isolation and purification of TfgL
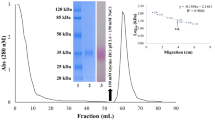
In present study, the lectin (TfgL) was purified from the seeds of Trigonella foenum-graecum (Fenugreek) by ammonium sulfate precipitation, anion-exchange, and size exclusion chromatography. Hemagglutination assays showed that crude extracts of Fenugreek seeds contain a lectin activity. By this observation, we designed a purification procedure in the present study which is simpler and well-organized for separating lectins by the fractionation of crude extract with 60% ammonium sulfate precipitation increased specific activity to 138.97 from 54.98 HU/mg. This ammonium sulfate precipitated soluble protein fraction was again fractionated on an anion exchange column (DEAE-sepharose) which was pre-equilibrated with 20 mM phosphate buffer, pH 7.6. Under these conditions, the majority of the proteins were eluted in the equilibration buffer in between 20–40 ml (peak D1). The fractions (peak D2) eluted at 80 ml of equilibration buffer showed specific activity 658.15 HU/mg (Fig. 1A). Peak D2 fractions were pooled and further fractionated by size exclusion chromatography on Sephadex G-50 into different peaks (Fig. 1B). Hemagglutination activity was detected highest (Specific activity 780.64 HU/mg) in the fraction P4. This specific activity is better than some other purified lectins (Li et al. 2012; Rego et al. 2002). The yield of TfgL is 25.3 mg from 30 g seeds. The purified TfgL represented a 14.2-fold increase in hemagglutinating activity over the crude extract, with overall activity and recovery of the protein approximately 19.1 and 1.35%, correspondingly (Table 1). The TfgL showed specific activity 780.64 HU per mg.

FPLC elution profiles for the fenugreek seed extracts. Ammonium sulfate precipitate was fractionated by ion exchange on DEAE-Sepharose (A) from where The unabsorbed fraction (D2) demonstrating hemagglutination activity was then separated by gel filtration on Sephadex G-50 (B)
Molecular mass and pI determination
The molecular mass of purified TfgL was measured by SDS-PAGE (Fig. 2A), which revealed a molecular weight of 27 kDa. The isoelectric point (pI) of the TfgL was determined as 6.3 and 6.7, based on the linear pH range 3–10 of isoelectric focusing performed by two-dimensional electrophoresis shown in Fig. 2B. These results reveal that the presence of two isolectins and suggests charge heterogeneity between the subunits. The acidic pIs may be attributed to the acidic nature of TfgL. These results resemble with the previously reported lectins (Konozy 2003).

A SDS–PAGE profile of TfgL: Lane 1. Molecular mass markers, lane 2. Purified TfgL. B Two-dimensional gel electrophoresis map of purified TfgL. In this case IEF was performed on linear pH 3‒10 IPG strips
MALDI-TOF-MS analysis
The peptide mass fingerprinting (PMF) of the 1-D SDS-PAGE gel bands of purified TfgL was obtained by MALDI-TOF–MS using α-cyano-4-hydroxycinnamic acid as matrix. The MALDI-TOF–MS spectrum was generated from in-gel trypsin digestion and is displayed in Fig. 3. The PMF peaks of TfgL are summarized in Table 2, after excluding the control spectrum produced by trypsin autolysis products. In addition, analyses by the MASCOT search program in NCBInr database revealed that the sequence is homologous with lectin. The observed monoisotopic mass of tryptically digested peptides obtained by MALDI-TOF–MS, their matched peptides sequence are presented in the Table 3.

MALDI-TOF–MS spectra (peptide mass fingerprints) of peptides generated from tryptically digested TfgL
Hemagglutination activity of TfgL
The hemagglutination activity of TfgL was stable in the temperature range 37–90 °C and at pH values 3, 7.6 and 10 (Fig. 4). The purified TfgL retained its activity greater than 95% up to 90 °C and also stable towards broad range of pHs but activity loss was greater at lower pH 3 than compared with higher pH 10. In contrast to other plant lectins reported in literature, TfgL was found to be relatively stable. The hemagglutination activity of black turtle bean lectin (He et al. 2014) and Zihua Snap Bean lectin (Jiang et al. 2019) were stable at temperatures up to 70 °C, and is reduced to half at 80 °C but haricot bean agglutinin is stable at temperatures up to 40 °C and is totally abolished at 80 °C. TfgL showed sensitivity towards acidic pH. This lectin activity remained almost unaffected from pH 4 to pH 10. On the other hand, it activity decreases at pH values below pH 4. This is resembles with EspecL, which is isolated from Erythrina speciosa seeds and the lectin of P. filicina were acidic pH sensitive (Sampaio et al 1998). But this is in contrast with the Phaseolus vulgaris L lectin, showed extremely stable in the acidic pH range of 2–7.5 (Sun et al. 2020).

Stability of TfgL. A Temperature stability of TfgL after incubation for 15 min at pH 7.6 in the range of 37–90 °C. B pH stability of TfgL at pHs 3, 7.6 and 10 after incubation for 30 min at 25 °C. Bar indicates standard deviation from triplicate determination. P ≤ 0.05 was considered statistically significant
Structural analysis of TfgL
Figure 5A displays the far‒UV CD curve for the native state of TfgL with minima, at 222 nm, which is characteristic of α-helical proteins. The Table 4 shows the different parts of secondary structure elements for this protein at different conditions. The values obtained for the various types of secondary structures are: α-helix (84.5%), β-sheet (6.5%), β-turns (5%) and unordered structures (4%) performed using CDNN software. The high α-helical content in the secondary structure of TfgL is very atypical for a legume lectin. The legume lectins identified so far contain predominantly β-sheet structure and have negligible or no α-helical content (Sultan et al. 2004a, b) but TfgL protein shows resemblance with the lectin DLL-II isolated from Dolichos lablab seeds which contains predominantly α-helical structure (Sultan et al. 2006).

A Effect of temperature wide range 20 to 90 °C length at 0.8 mg/ml protein concentration in 50 mM sodium phosphate buffer, pH 7.0, B effect of pH 3, 7.6 and 10and C effect of different concentrations of Gdn.HCl (0‒4 M) in20 mM phosphate buffer, pH 7.6,on the secondary structure of TfgL
CD studies at increasing temperature reveals that the backbone protein folding with no considerable change in CD spectra up to 90 °C demonstrated the thermostability of TfgL structure (Fig. 5A). The effect of pH studies showed that structure of TfgL does not alter over a wide range of pH, even though some loss was observed at acidic pH 3 which correlates with hemagglutination activity (Fig. 5B).
Gdn-HCl, which is a strong chemical denaturant, was utilized to examine whether this compound could have effect on secondary structure of TfgL(Fig. 5C). The stability of TfgL is reflected by the low level of structure disruption showed at 1.5 M Gdn-HCl. At 1.5‒3.5 M Gdn-HCl concentrations, a significant reduction of the normal conformations takes place revealing that TfgL does not conserve its structure at these denaturant concentrations. Native to unfolded state transition takes place between 1.5 to 3.5 M concentration of Gdn-HCl, with the midpoint of transition (Tm) being approximately 2.5 M Gdn-HCl. At 4.0 M Gdn-HCl, the protein lost 54% of secondary structure and wholly unfolded at 6.0 M Gdn-HCl. In order to investigate the reversibility of chemical unfolding of TfgL, samples of the protein pre-incubated with 6 M Gdn.HCl were diluted to different concentrations by dialysis, then the changes in the CD spectra properties were measured (Data were not shown). The results indicated that complete chemical denaturation of TfgL shows irreversible process. The different components of secondary structure elements for this protein at different conditions showed that α-helical content was retained in all extreme conditions but in presence of denaturant like Gdn.HCl this α-helical structure changes to Random coil and β-turn.
Fluorescence spectra of TfgL in the presence of different quenchers in various concentrations revealed that there is a progressive reduction in the emission intensity upon raising the concentration of the quenchers. Addition of iodide ion (I‒) (charged) quencher showed much lower quenching in contrast to acrylamide (neutral quenchers). The degree of quenching achieved in each case, at a resultant quencher concentration of 0.6 M is shown in Table 5. The fluorescence emission properties of tryptophan (indole side-chain residue) in proteins are extremely responsive to the surrounding environment and consequently are very frequently employed to examine the changes in conformation and structure that takes place in proteins under different conditions (Eftink and Ghiron 1981). Trytophan residues which are in non-polar environment shows the fluorescence around 320 nm, while those that are in a polar environment produce at a significantly greater wavelength. The emission maximum of TfgL native protein at 327 nm evidently indicates that these residues are mostly buried in the hydrophobic interior of the protein. Red shift to 348 nm is observed upon denaturation with 6 M Gdn-HCl (spectra not shown).
The Stern–Volmer plots achieved with different quenchers are displayed in Fig. 6A. The quenching pattern obtained for TfgL with acrylamide and iodide ion showed a linear dependence on the concentration of quencher, from the slopes of which, the Stern–Volmer quenching constants were calculated Eq. (3). The values obtained are listed in Table 5. The modified Stern–Volmer plots obtained with two quenchers are given in Fig. 6B. From the Y-intercepts of these plots, fa, the value of the fraction of accessible Tryptophan residues in each case, was determined and the value of the corresponding quenching constant, Ka was calculated using Eq. (4). These values are also shown in Table 5. It can be clearly seen from the data presented that, in TfgL about 66% of the fluorescence intensity was accessible to acrylamide, while only 28% of the fluorescence intensity was accessible iodide ion (Table 5). The value of Ka for acrylamide was 5.50 M−1 and for iodide it was 3.45 M−1, clearly indicating the acrylamide to be a much more efficient quencher than iodide ion. These results show similarity to earlier fluorescence studies on lectins. The considerably lower quenching and accessibility by the charged quencher I‒ are consistent with their incapability to enter into the protein hydrophobic core. This provides additional support to the above elucidation made from the study of emission maximum, that the Trp residues of TfgL are buried in the protein core which is mostly hydrophobic. These results show similarity to earlier fluorescence studies on lectins (Sultan et al. 2006).

A Stern‒Volmer and B Modified Stern‒Volmer plots of fluorescence quenching data for TfgL. Quenching profile with acrylamide (■) and iodide ion (●). The emission spectra of the protein solutions (in 10 mM phosphate buffer, pH 7.0) were measured in the region of 310‒440 nm at 25 °C. All samples were centrifuged before use and the clear supernatant was used for experiments
Porphyrin binding to TfgL
Absorption spectra in the Soret band region for MnTSPP, a tetra-anionic porphyrin and for MnTMPyP, a tetra-cationic porphyrin, in the absence and presence of TfgL, are shown in Fig. 7A, B, respectively. In each figure, spectrum 1 corresponds to the porphyrin alone and remaining spectra correspond to those obtained in the presence of increasing concentrations of TfgL (Fig. 7E, F). This plot clearly indicates that the data has a linear dependence. From the X-intercept of this plot, the association constant, Ka, for the association of MnTSPP and MnTMPyP with TfgL was estimated to be 1.2 × 106 M‒1 and 3.45 × 106 M‒1 respectively. Studies carried out in the last decade indicate that porphyrins bind to different plant lectins with considerable affinity (Komath et al. 2006). The analysis of TfgL binding with MnTMPyP and MnTSPP reveals that high binding affinity than those observed for the other lectin-porphyrin systems (Kenoth et al. 2001). This result reveals that TfgL showed high binding affinity towards MnTMPyP compared with MnTSPP and this may due to the acidic nature of TfgL.

Absorption spectra of porphyrins in the absence and in the presence of TfgLin 20 mM sodium phosphate buffer, pH 7.4. Porphyrin samples (2 µM) were titrated by addition of small aliquots of the TfgL. A MnTSPP, B MnTMPyP. Binding curve for the interaction of C MnTSPP, D MnTMPyP with TfgL. Chipman plot for E MnTSPP, F MnTMPyP binding to TfgL
In conclusion, we are purified and characterized lectin (TfgL) from Trigonella foenum-graecum (fenugreek) seeds. TfgL showed high α-helical content, and also showed hemagglutination activity. The secondary structure of this protein stability towards temperature, pH and Gdn.HCl has been investigated. We have investigated the tryptophan exposure and environment which revealed that the Trp residues are partially buried. TfgL has high binding affinity towards porphyrins.
Abbreviations
- TfgL:
-
Trigonella foenum-graecum lectin
- 2D-PAGE:
-
Two-dimensional gel electrophoresis
- MALDI-TOF–MS:
-
Matrix assisted laser desorption ionization-time of flight mass spectrometry
- Gdn.HCl:
-
Guanidine hydrochloride
- CD:
-
Circular dichroism
- MnTSPP:
-
5,10,15,20-Tetrakis(4-sulfonatophenyl)-21H,23H-porphine manganese(III) chloride
- MnTMPyP:
-
Manganese(III) 5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine chloride tetrakis (methochloride)
References
Blum H, Beier H, Gross HJ (1987) Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis 8:93–99
Cavada BS, Bari AU, Pinto-Junior VR, Lossio CF, Silva MTL, Souza LAG, Oliveira MV, Souza-Filho CHD, Correia SEG, Vital APMS et al (2020) Purification and partial characterization of a new lectin from parkia panurensis benth. Ex H.C. Hopkins seeds (leguminosae family; mimosoideae subfamily) and evaluation of its biological effects. Int J Biol Macromol 145:845–855
da Silva JDF, da Silva SP, da Silva PM, Vieira AM, de Araujo LCC, de Albuquerque LT, de Oliveira APS, do Nascimento Carvalho LV, da Rocha Pitta MG, de Melo Rego MJB (2019) Portulaca elatior root contains a trehalose-binding lectin with antibacterial and antifungal activities. Int J Biol Macromol 126:291–297
Eftink MR, Ghiron CA (1981) Fluorescence quenching studies with proteins. Anal Biochem 114:199–227
He S, Shi J, Ma Y, Xue SJ, Zhang H, Zhao S (2014) Kinetics for the thermal stability of lectin from black turtle bean. J Food Engg 142:132–137
Jiang B, Wang X, Wang L, Lv X, Li D, Liu C, Feng Z (2019) Two-step isolation, purification, and characterization of lectin from zihua snap bean (Phaseolus vulgaris) Seeds. Polymers 11:785
Kenoth R, Reddy DR, Maiya BG, Swamy MJ (2001) Thermodynamic and kinetic analysis of porphyrin binding to Trichosanthes cucumerina seed lectin. Eur J Biochem 268:5541–5549
Komath SS, Kavitha M, Swamy MJ (2006) Beyond carbohydrate binding: new directions in plant lectin research. Org Biomol Chem 4:973–988
Konozy EHE, Bernardes ES, Rosa C, Faca V, Greene LJ, Ward RJ (2003) Isolation, purification, and physicochemical characterization of a D-galactose-binding lectin from seeds of Erythrina speciosa. Arch Biochem Biophys 410:222–229
Kou J, Dou D, Yang L (2017) Porphyrin photosensitizers in photodynamic therapy and its applications. Oncotarget 8:81591–81603
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lagarda-Diaz I, Guzman-Partida A, Vazquez-Moreno L (2017) Legume lectins: proteins with diverse applications. Int J Mol Sci 18:1242
Li T, Yin X, Liu D, Ma X, Lv H, Sun S (2012) Isolation and characterization of a novel lectin with antifungal and antiproliferative activities from Sophora alopecuroides seeds. Acta Biochim Biophys Sin (shanghai) 44:606–613
Liang X, Chen M, Bhattarai P, Hameed S, Dai Z (2020) Perfluorocarbon @ Porphyrin nanoparticles for tumor hypoxia relief to enhance photodynamic therapy against liver metastasis of colon cancer. ACS Nano 14:13569–13583
Mishra A, Behura A, Mawatwal S, Kumar A, Naik L, Mohanty SS, Manna D, Dokania P, Mishra A, Patra SK, Dhiman R (2019) Structure-function and application of plant lectins in disease biology and immunity. Food Chem Toxicol 134:110827-110844.
Oddepally R, Guruprasad L (2015) Isolation, purification, and characterization of a stable defensin-like antifungal peptide from Trigonella foenum-graecum (fenugreek) seeds. Biochem Mosc 80:332–342
Oddepally R, Sriram G, Guruprasad L (2013) Purification and characterization of a stable Kunitz trypsin inhibitor from Trigonella foenum-graecum (fenugreek) seeds. Phytochemistry 96:26–36
Rego EJ, de Carvalho DD, Marangoni S, de Oliveira B, Novello JC (2002) Lectins from seeds of Crotalaria pallida (smooth rattlebox). Phytochemistry 60:441–446
Sampaio AH, Rogers DSJ, Barwell CJ (1998) A galactose-specific lectin from the red marine alga, Ptilota filicina. Phytochem 48:765–769
Sitohy M, Doheim M, Badr H (2007) Isolation and characterization of a lectin with antifungal activity from Egyptian Pisum sativum seeds. Food Chem 104:971–979
Srinivasan K (2006) Fenugreek (Trigonella foenum-graecum): a review of health beneficial physiological effects. Food Rev Int 22:203–224
Sultan NA, Kenoth R, Swamy MJ (2004a) Purification, physicochemical characterization, saccharide specificity, and chemical modification of a specific lectin from the seeds of Trichosanthes dioica. Arch Biochem Biophys 432:212–221
Sultan NA, Maiya BG, Swamy MJ (2004b) Thermodynamic analysis of porphyrin binding to Momordica charantia (bitter gourd) lectin. Eur J Biochem 271:3274–3282
Sultan NA, Rao RN, Nadimpalli SK, Swamy MJ (2006) Tryptophan environment, secondary structure and thermal unfolding of the galactose-specific seed lectin from Dolichos lablab: fluorescence and circular dichroism spectroscopic studies. Biochim Biophys Acta 1760:1001–1008
Sun X, He S, Ye Y, Cao X, Liu H, Wu Z, Yue J, Jin R, Sun H (2020) Combined effects of pH and thermal treatments on IgE-binding capacity and conformational structures of lectin from black kidney bean (Phaseolus vulgaris L). Food Chem 329:127183
Tsaneva M, Van Damme JM (2020) 130 years of plant lectin research. Glycoconjugate J 37:533–551
Acknowledgements
We are grateful to Prof. Lalitha Guruprasad School of Chemistry, University of Hyderabad for providing theinfrastructure. We also thank School of Chemistry, University of Hyderabad for providing various equipments for the project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rajender, O., Pallavi, H. & Sultana, R. Purification and characterization of a lectin from Trigonella foenum-graecum (fenugreek) seeds and its porphyrin binding studies. J. Plant Biochem. Biotechnol. 33, 341–352 (2024). https://doi.org/10.1007/s13562-024-00894-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13562-024-00894-0