
Abstract
Analysis of global protein expression, an approach known as expression proteomics, can offer important clues for understanding tumor biology that cannot be obtained by other approaches (e.g., genome or transcriptome analysis). Using two-dimensional difference gel electrophoresis (2D-DIGE) and mass spectrometry, we performed global protein expression studies of bone and soft tissue sarcomas to develop novel diagnostic and therapeutic biomarkers and allow molecular classification of the tumors. Among 1500 protein variants identified in the two-dimensional gel, 67 proteins correctly distinguished the eight subtypes of 99 histologically classified soft tissue sarcomas. Hierarchical clustering demonstrated leiomyosarcoma and MFH shared a similar protein expression profile, and clear cell sarcoma, synovial sarcoma, and MPNST could be grouped according to their protein expression patterns. Pleomorphic leiomyosarcoma and MFH showed similar tropomyosin isoform expression patterns. Patients with gastrointestinal stromal tumors expressing pfetin protein had better survival than those whose tumors lacked it. We identified 10 protein spots associated with the chemosensitivity of osteosarcoma to preoperative chemotherapy. These 10 spots could be new diagnostic and prognostic markers for osteosarcoma and new therapeutic targets for the disease. Proteomic analysis using 2D-DIGE provides novel information on the biology of bone and soft tissue sarcomas that could be used to diagnosis and treat these tumors.
Level of Evidence: Level II, diagnostic study. See the Guidelines for Authors for a complete description of levels of evidence.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past three decades, advances in diagnostic modalities and treatment methods have substantially improved the survival rate and postoperative limb function of patients with bone and soft tissue sarcomas [5, 10, 20, 27, 40]. Wide excision of tumors in conjunction with multiagent chemotherapy now provides 5-year disease-specific survivals ranging from 60% to 80% in patients with localized high-grade sarcomas [2, 5, 27]. Despite this success, the outcomes of patients who have metastatic disease at diagnosis or those with tumors showing a poor response to chemotherapy are still unsatisfactory (5-year disease-specific survival rates, 20%–40%), even with dose-intensive or high-dose chemotherapy [9, 18, 19, 33]. For improving the prognosis of patients with these difficult-to-treat sarcomas, it is imperative to develop new targeted therapeutic strategies based on an understanding of the biologic mechanisms underlying the metastasis and chemoresistance of these tumors.
Recent development of high-throughput screening techniques has allowed global investigations of the molecular backgrounds associated with the clinicopathologic characteristics of tumors. A DNA microarray-based approach allows the screening of several thousand mRNAs in bone and soft tissue sarcomas and can identify the genes relevant to their histologic diagnosis, clinical features, and chemosensitivity [39]. Using an oligonucleotide microarray approach, Nakayama et al. [31] analyzed gene expression in 105 soft tissue sarcoma samples and reclassified malignant fibrous histiocytoma (MFH), which has a wide variety of clinicopathologic features, into pleomorphic subtypes of other distinct types of sarcomas. Based on gene-expression profiles, Ochi et al. [34] identified 60 genes whose expression levels were likely correlated with the chemosensitivity of osteosarcomas. Mintz et al. [29] reported 104 genes were differentially expressed between chemotherapy-sensitive and -resistant osteosarcomas.
Although much evidence suggests genetic abnormalities play a primary role in the development of tumors, there is also evidence for the effects of aberrations that cannot be detected solely by genome (DNA sequencing) or transcriptome (measurement of all mRNA in a population of cells) analysis: posttranslational modifications of proteins such as phosphorylation, glycosylation, and degradation, are aberrantly regulated in many types of cancers and cannot be predicted by DNA sequencing or measurement of mRNA expression. It has become evident there is considerable discrepancy between expression of mRNA and that of protein [4, 12]. Further, proteins are more directly linked to aberrant tumor phenotypes. These difficulties underline the potential advantages of monitoring protein expression in a global manner, an approach known as expression proteomics. In addition, the results obtained from proteomic studies will be more easily applicable to the clinical field with the use of specific antibodies.
We have performed global protein expression studies using our original high-throughput two-dimensional difference gel electrophoresis (2D-DIGE) system on several types of tumors [21, 22]. This approach has proven useful for classifying soft tissue sarcomas at the protein expression level and for identifying novel diagnostic and prognostic markers [37]. Biomarkers specifically expressed in a specific subgroup of tumors can facilitate a risk-adopted tailored medical treatment. Examination of the markers may allow the identification of a high-risk patient group that may benefit from adjuvant therapy, whereas it may also help spare low-risk patients unnecessary treatment. Moreover, specifically expressed molecules or associated pathways could be crucial molecular targets to allow for more selective therapeutic intervention.
We report our approach for identifying biomarkers that associate with clinicopathologic features of bone and soft tissue sarcomas using global protein expression analysis.
Materials and Methods
We performed protein expression profiling for 99 soft tissue sarcomas: 28 MFHs, 19 rhabdomyosarcomas, 12 synovial sarcomas, 10 leiomyosarcomas, 10 myxoid liposarcomas, nine gastrointestinal stromal tumors (GIST), six clear cell sarcomas, and five malignant peripheral nerve sheath tumors (MPNST) using 2D-DIGE. We used the term “MFH” to describe tumors diagnosed as storiform and pleomorphic-type MFH that showed predominant pleomorphic features without any findings of a specific type of differentiation. All tumor samples were obtained from patients treated at the National Cancer Center Hospital (Tokyo, Japan). We snap-froze fresh-frozen samples in liquid nitrogen immediately after resection or biopsy sampling before any treatment and stored them at −80°C until use. Tumor samples for immunohistochemical analysis were preserved in archival paraffin-embedded tissue blocks. This study was approved by the Institutional Review Board and conducted according to tenets of the Declaration of Helsinki.
Protein expression profiles were studied by 2D-DIGE as described previously [21, 22]. For protein extraction, the frozen samples were homogenized with urea lysis buffer (6 M urea, 2 M thiourea, 3% CHAPS, 1% Triton X-100). After centrifugation at 15,000 rpm for 30 minutes, the supernatant was used for studies. A mixture of all the experimental samples was used as an internal control sample. The internal control and individual samples were labeled with fluorescent dyes with different excitation and emission wavelengths (CyDye DIGE Fluor saturation dye; Amersham Biosciences, Little Chalfont, Buckinghamshire, UK): Cy3 for the internal control and Cy5 for the individual samples. The Cy3-labeled control sample and the Cy5-labeled individual sample were mixed and coseparated by two-dimensional polyacrylamide gel electrophoresis. The first-dimensional separation was performed using an immobilized pH gradient gel (length 24 cm, pI range 3-10; Amersham Biosciences) in accordance with the manufacturer’s recommended protocol. The immobilized pH gradient gels were then equilibrated and transferred to 9% to 16% polyacrylamide gradient gels measuring 24 cm × 20 cm. The gels were run in an Ettan Dalt II system (Amersham Biosciences) at 18 W and 18°C for 15 hours. Each sample was run on triplicate gels and the average spot intensities were calculated for quantitative analysis.
After electrophoresis, the gels were scanned at appropriate wavelengths for Cy3 and Cy5 using a 2-D 2920 MasterImager (Amersham Biosciences) [21, 22]. The ratio of Cy5 intensity to Cy3 intensity was calculated for all spots in each gel with DeCyder software (Amersham Biosciences) to obtain the standardized spot intensities. Because the Cy3 image represents the internal standard sample, this standardization procedure eliminates gel-to-gel differences. Approximately 1500 protein spots were visualized by laser scanning (Fig. 1). The standardized spot intensities were logarithmically transformed and analyzed with the data-mining package Impressionist (GeneData, Basel, Switzerland).

Representative two-dimensional image of Cy5-labeled proteins of osteosarcoma. Approximately 1500 protein spots are visualized by laser scanning. Proteins corresponding to the spots of interest were picked up and identified by mass spectrometric analysis.
We identified informative proteins using a support vector machine algorithm with leave-one-out crossvalidation. The classification performance of a candidate classifier was evaluated by multivariate analysis, including principal component analysis and hierarchical clustering. Proteins corresponding to the spots of interest were identified by mass spectrometric analysis using a matrix laser desorption/absorption/ionization (MALDI) time of flight mass spectrometer. Protein identification and differential expression were confirmed by Western blotting using specific antibodies.
Immunohistochemical analysis was performed according to the streptavidin-biotin peroxidase method using a Strept ABC Complex/HRP kit (DAKO, Tokyo, Japan) on formalin-fixed paraffin-embedded tissues. Rabbit polyclonal antibody against pfetin was kindly provided by Dr Morton [35]. Two reviewers (YS, AK) reviewed the results of immunohistochemical staining in a blinded fashion regarding the clinical data.
We calculated tumor-specific survival time from the first resection of primary tumor to death from tumor-specific causes. Patients who died of causes unrelated to the disease were censored at the time of death. All time to event end points were computed using the Kaplan and Meier method. We used the log-rank test to determine differences in survival between subgroups. We used the Cox proportional hazards regression to analysis differences in survival after adjusting for potentially confounding variables (site, size, differentiation and pfetin).
To identify markers associated with osteosarcoma chemosensitivity, we analyzed the protein expression profiles of 23 biopsy samples of typical osteosarcoma (conventional central high-grade osteosarcomas that had developed in long tubular bones in patients with a mean age of 18 years). All the patients received standard pre- and postoperative chemotherapy consisting of high-dose methotrexate, cisplatin, doxorubicin, and ifosfamide. The histologic effects of preoperative chemotherapy according to the Huvos grading system [36] were Grade I in seven cases, Grade II in seven, Grade III in six, and Grade IV in three. We compared the protein expression profiles of two distinct groups; nine samples from patients showing a good response to preoperative chemotherapy (greater than 90% tumor necrosis; Huvos Grades III and IV) and seven samples from patients showing a poor response (less than 50% tumor necrosis; Huvos Grade I).
Results
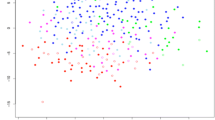
Hierarchical clustering using all 1500 protein spots did not classify the tumors exactly in accordance with the histologic classification. To examine the possible differences in protein expression between different types of sarcoma, we selected 67 proteins whose expression was associated with the existing histologic classification by using a support vector machine algorithm (Fig. 2). All the tumors were separable into six clusters. Myxoid liposarcomas and rhabdomyosarcomas formed tight clusters on two distinct dendrogram branches. The third and fourth clusters mostly comprised a mixture of leiomyosarcomas and MFHs. The third cluster contained a large proportion of leiomyosarcomas, whereas the fourth cluster contained mostly MFHs together with two cases of pleomorphic leiomyosarcoma and a case of CCS. GISTs formed a distinct fifth cluster. The sixth cluster comprised three different types of sarcomas (CCS, synovial sarcoma, and MPNST) and three outlier cases.

Hierarchical classification of the 99 soft tissue sarcomas using the selected 67 proteins. The dendrogram shows the degree of similarity of protein expression pattern between the tumors. The shorter the branches, the more similar the two joined tumors. Most tumors formed tight clusters corresponding to their histology except leiomyosarcoma and malignant fibrous histiocytoma. RMS = rhabdomyosarcoma; MFH = malignant fibrous histiocytoma; GIST = gastrointestinal stromal tumor; MPNST = malignant peripheral nerve sheath tumor.
Using supervised analysis of pleomorphic and conventional leiomyosarcomas, we found tropomyosin isoforms were responsible for leiomyosarcoma subtypes. Conventional leiomyosarcoma expressed mainly tropomyosin isoforms 1 and 2, whereas pleomorphic leiomyosarcoma largely expressed tropomyosin isoforms 3 and 4 (Fig. 3). MFH shared an expression pattern of tropomyosin isoforms (mainly types 3 and 4) similar to that of pleomorphic leiomyosarcoma.

Immunohistochemical staining of tropomyosin isoform type 4. Representative photomicrographs of (A) conventional leiomyosarcoma and (B) pleomorphic leiomyosarcoma. Tropomyosin isoform 4 is highly expressed in pleomorphic leiomyosarcoma, but not in conventional leiomyosarcoma (stain, immunohistochemical staining for tropomyosin isoform 4; original magnification, ×200).
By comparing the protein expression profiles of two distinct groups of GISTs—pathologically high-risk and clinically aggressive cases and pathologically low- or intermediate-risk and clinically indolent cases—we identified 43 protein spots whose expression differed (Wilcoxon test, p < 0.001) between the two groups. Mass spectrometric identification demonstrated the 43 spots corresponded to 25 distinct gene products, eight of which were derived from a potassium channel protein, pfetin. Western blotting and real-time PCR suggested pfetin expression inversely correlated (p < 0.0001) with the development of metastasis. Immunohistochemical analysis revealed patients with tumors expressing pfetin protein had better (p < 0.0001) tumor-specific survival than those with tumors lacking pfetin expression (Fig. 4).

Tumor-specific survival of 210 patients with M0 gastrointestinal stromal tumors according to the expression of pfetin protein in the primary tumor samples examined by immunohistochemistry. Patients with tumors expressing pfetin (n = 171) had better (p < 0.0001) survival (5-year survival, 97.2%) than those with tumors lacking pfetin expression (n = 39) (5-year survival, 76.5%).
By comparing the protein expression profiles (1465 protein spots) of two distinct groups of osteosarcoma biopsy samples—a chemosensitive group (good responders; Huvos Grades III and IV) versus a chemoresistant group (poor responders; Huvos Grade I)—we identified 10 protein spots whose expression differed (Wilcoxon test, p < 0.05) between the two groups (Fig. 5). Among these 10 spots, the expression levels of seven spots were higher and those of three spots were lower in poor responders compared with good responders. Principal component analysis accurately divided the osteosarcoma samples into good responder and poor responder groups based on the expression levels of the 10 selected classifiers (Fig. 6).

Expression pattern of the 10 protein spots that discriminate good responders (Huvos Grades III, IV) from poor responders (Huvos Grade I) among 16 patients with osteosarcoma. Horizontal rows represent individual protein spots, and vertical columns represent individual samples. Each cell in the matrix represents the expression level of a single protein spot in a single sample. Red indicates overexpression relative to the expression of the control sample, whereas green indicates underexpression. There are seven spots that are overexpressed in poor responders and three spots overexpressed in good responders.

Principal component analysis of the 16 osteosarcomas on the basis of the expression profiles of the 10 selected protein spots. It accurately classifies the osteosarcoma samples into either good responders (Huvos Grades III, IV) or poor responders (Huvos Grade I).
Discussion
Many lines of evidence have revealed genetic alterations such as activation of oncogenes, inactivation of tumor suppressor genes, and chromosomal translocations play a primary role in the development of bone and soft tissue sarcomas [6, 16, 24]. These genetic alterations initiate changes in a series of signal transduction pathways and develop malignant tumor cells. Although the initial changes occur at the DNA level, all the effects are ultimately expressed as the changes of the protein content. Proteins are considered the hallmark of genetic alterations. Global mRNA expression studies are often performed to follow these changes [29, 30–34]. However, recent comparative studies of mRNA and protein expression in tumor cells revealed only a subset of mRNA correlated with protein abundance [4, 12]. Moreover, posttranslational modifications, which play important roles in tumor biology, cannot be predicted only by mRNA examinations. These limitations of mRNA expression studies underline the potential advantages of monitoring protein expression in a global manner, an approach known as expression proteomics. Recently, using MALDI MS tissue profiling, Holt et al. [14] reported protein peaks that were differentially expressed in high-grade and low-grade soft tissue sarcomas. Among several proteomic technologies, 2D-DIGE has advantages that make it possible to examine hundreds of proteins simultaneously estimating their expression and posttranslational modifications quantitatively [21, 22]. Applications of sensitive fluorescent dyes and laser scanning made it possible to execute large-scale proteomics in a more efficient, accurate, and reproducible way. In the present study, we examined the protein expression profiles of a diverse group of bone and soft tissue sarcomas in relation to their clinicopathologic features using the 2D-DIGE technique.
A limitation of the current 2D-DIGE is the thoroughness of the analysis. Although the method reveals global expression profiles of more than 1000 protein spots quantitatively, it cannot uncover proteins expressed in very low levels. Compared with the cDNA microarray analysis (20,000 probe sets), the sensitivity of the current 2D-DIGE analysis (1500 spots) is unsatisfactory. We are now trying to improve the sensitivity of 2D-DIGE using a large-format polyacrylamide gel (24 cm × 38 cm) for two-dimensional polyacrylamide gel electrophoresis. Preliminary results demonstrated more protein spots (5000 spots) are quantitatively detected in the large-format two-dimensional gels. We believe such efforts may overcome some of the inherent limitations of the proteomics.
We found 67 proteins correctly distinguished the eight subtypes of 99 soft tissue sarcomas according to their histologic classification. Hierarchical clustering demonstrated leiomyosarcoma and MFH shared a similar protein expression profile, some of which was not distinguishable on the basis of protein expression, and that clear cell sarcoma, synovial sarcoma, and MPNST were grouped according to their protein expression patterns. These results were generally consistent with previous reports based on histologic and transcriptome analyses [7, 13, 19, 25, 30–32].
Using immunohistochemical analysis, Hasegawa et al. [13] demonstrated a large subset of MFH expressed poorly differentiated smooth muscle or myofibroblastic features and should be regarded as pleomorphic leiomyosarcoma or pleomorphic myofibrosarcoma. Fletcher et al. [7] examined 100 cases of so-called MFH histologically and reclassified 20 of the cases as leiomyosarcomas. Considering these findings together with recent DNA microarray and comparative genomic hybridization analyses [25, 31, 32], at least a proportion of MFHs should be reclassified as a pleomorphic subtype of leiomyosarcoma. In this context, pleomorphic leiomyosarcoma and MFH shared similar expression patterns of tropomyosin isoforms.
The similarity of the protein expression profiles of clear cell sarcoma, synovial sarcoma, and MPNST is consistent with the biologic associations and similar gene expression patterns of these tumors. Clear cell sarcoma is also known as melanoma of soft parts and is derived from neural crest progenitor cells [19]. Nagayama et al. [30] reported synovial sarcoma and MPNST shared similar expression patterns of numerous genes related to neural differentiation and suggested synovial sarcoma might be of neuroectodermal origin. On the basis of protein expression patterns, our present results appear to support the assumption that these three tumors are derived from (or differentiate to) neural or neuroectodermal cells.
Currently, the therapeutic strategy for bone and soft tissue sarcomas largely depends on the histologic subtype, grade, and stage of the disease [11, 26]. Although the systems provide valuable information about the clinical behavior of the tumors in most cases, there are several outliers (or patients) in which their clinical course could not be predicted only on the basis of classic appraisals. Moreover, the introduction of molecular-targeted drugs has changed the situation dramatically [8, 28, 41]. Most of these new drugs target a specific molecule, whose presence could not be predicted only from the classic clinicopathologic features. Novel clues for identifying prognostic markers and therapeutic targets in individual tumors are urgently required.
GISTs are the most common mesenchymal tumors of the gastrointestinal tract and are characterized by a wide spectrum of clinical outcomes [41]. Identification of a high-risk group of patients who may benefit from adjuvant therapy such as that including imatinib is an urgent clinical concern. We found 43 proteins, including eight variants of pfetin, which was originally reported as a protein highly expressed in fetal cochlea and brain, were correlated with the outcome of patients with GIST [35, 38]. Although the expression of a whole 43-protein set predicted the outcome of GIST correctly, it seemed difficult to apply the results directly to a hospital setting. Cutting-edge genome or proteome technologies cannot be transferred easily to ordinary clinical examinations. A smaller number of targets, measurable by simpler methods such as immunohistochemistry, is preferable for use in practice. We found expression of pfetin protein, which could be examined immunohistochemically using paraffin-embedded archival tissues, predicted (p < 0.0001) disease-specific survival in patients with GIST [38]. Examination of pfetin expression using immunohistochemistry may allow the identification of high-risk patients with GIST who may benefit from adjuvant treatment such as that including imatinib and minimize the risk of unnecessary treatment for low-risk patients.
The prognosis of patients with osteosarcoma has improved markedly as a result of effective chemotherapy. Dose-intensive multiagent chemotherapy continued for 1 year or so has resulted in long-term survival for the majority of patients with localized osteosarcoma [27]. Despite this success, however, 30% to 40% of patients, mainly those with tumors showing a poor response to preoperative chemotherapy, will experience relapse, most often with pulmonary metastases, and die [2]. Prediction of chemosensitivity at the time of diagnosis would therefore be of great clinical importance. Over the last decade, considerable efforts have been made for this purpose. Not only classic parameters such as histologic subtype, tumor location, and presence of metastatic disease at diagnosis [1], but also several molecules related to drug metabolism/transport or tumor development have been reported to correlate with the chemosensitivity of osteosarcoma [3, 15, 17, 23].
Based on the results of cDNA microarray consisting of 23,040 genes, Ochi et al. [34] identified 60 genes whose expression was likely correlated with the chemosensitivity of osteosarcoma and proposed a scoring system based on the expression levels of these genes. Comprehensive analysis of expression levels among thousands of genes enables investigators to identify novel important molecules that cannot be predicted from conventional data. Mintz et al. [29] reported osteosarcoma chemoresistance was associated with osteoclastogenesis and bone resorption based on decreased expression of osteoclastogenesis-inhibitory factors in tumors showing a poor response to chemotherapy. We report here 10 protein spots associated with the chemosensitivity (necrosis rate) of osteosarcoma to preoperative chemotherapy. Although the 10 spots are currently under investigation, further studies may lead to new diagnostic or prognostic markers for osteosarcoma and new therapeutic targets.
Proteomic analysis using 2D-DIGE can provide important, novel clues for understanding the biology of bone and soft tissue sarcomas and for revealing candidate tumor markers and therapeutic targets.
References
Bacci G, Ferrari S, Delepine N, Bertoni F, Picci P, Mercuri M, Bacchini P, Brach del Prever A, Tienghi A, Camandone A, Campanacci M. Predictive factors of histologic response to primary chemotherapy in osteosarcoma of the extremity: study of 272 patients preoperatively treated with high-dose methotrexate, doxorubicin, and cisplatin. J Clin Oncol. 1998;16:658-663.
Bacci G, Longhi A, Fagioli F, Briccoli A, Versari M, Picci P. Adjuvant and neoadjuvant chemotherapy for osteosarcoma of the extremities: 27 year experience at Rizzoli Institute, Italy. Eur J Cancer. 2005;41:2836–2845.
Baldini N, Scotlandi K, Barbanti-Brodano G, Manara MC, Maurici D, Bacci G, Bertoni F, Picci P, Sottili S, Campanacci M, Serra M. Expression of p-glycoprotein in high-grade osteosarcoma in relation to clinical outcome. N Engl J Med. 1995;333:1380–1385.
Chen G, Gharib TG, Huang CC, Taylor JMG, Misek DE, Kardia SLR, Giordano TJ, Iannettoni MD, Orringer MB, Hanash SM, Beer DG. Discordant protein and mRNA expression in lung adenocarcinomas. Mol Cell Proteomics. 2002;1:304–313.
Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin. 2004;54:94–109.
de Alava E. Molecular pathology in sarcomas. Clin Transl Oncol. 2007;9:130–144.
Fletcher CDM, Gustafson P, Rydholm A, Willen H, Akerman M. Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: prognostic relevance of subclassification. J Clin Oncol. 2001;19:3045–3050.
Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JVS, Booth BP, Verbois SL, Morse DE, Liang CY, Chidambaram N, Jiang JX, Tang S, Mahjoob K, Justice R, Pazdur R. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13:1367–1373.
Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJH, Pritchard DJ, Gebhardt MC, Dickman PS, Perlman EJ, Meyers PA, Donaldson SS, Moore S, Rausen AR, Vietti TJ, Miser JS. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694–701.
Grimer RJ. Surgical options for children with osteosarcoma. Lancet Oncol. 2005;6:85–92.
Guillou L, Coindre JM, Bonichon F, Bui NB, Terrier P, Collin F, Vilain MO, Mandard AM, Doussal VL, Leroux A, Jacquemier J, Duplay H, Sastre-Garau X, Costa J. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol. 1997;15:350–362.
Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730.
Hasegawa T, Hasegawa F, Hirose T, Sano T, Matsuno Y. Expression of smooth muscle markers in so called malignant fibrous histiocytomas. J Clin Pathol. 2003;56:666–671.
Holt GE, Schwartz HS, Caldwell RL. Proteomic profiling in musculoskeletal oncology by MALDI mass spectrometry. Clin Orthop Relat Res. 2006;450:105–110.
Ifergan I, Meller I, Issakov J, Assaraf YG. Reduced folate carrier protein expression in osteosarcoma: implications for the prediction of tumor chemosensitivity. Cancer. 2003;98:1958–1966.
Janknecht R. EWS-ETS oncoproteins: the linchpins of Ewing tumors. Gene. 2005;363:1–14.
Kakar S, Mihalov M, Chachlani NA, Ghosh L, Johnstone H. Correlation of c-fos, p53, and PCNA expression with treatment outcome in osteosarcoma. J Surg Oncol. 2000;73:125–126.
Kasper B, Lehnert T, Bernd L, Mechtersheimer G, Goldschmidt H, Ho AD, Egerer G. High-dose chemotherapy with autologous peripheral blood stem cell transplantation for bone and soft-tissue sarcomas. Bone Marrow Transplant. 2004;34:37–41.
Kawai A, Hosono A, Nakayama R, Matsumine A, Matsumoto S, Ueda T, Tsuchiya H, Beppu Y, Morioka H, Yabe H. Clear cell sarcoma of tendons and aponeuroses: a study of 75 patients. Cancer. 2007;109:109–116.
Kawai A, Muschler GF, Lane JM, Otis JC, Healey JH. Prosthetic knee replacement after resection of a malignant tumor of the distal part of the femur. J Bone Joint Surg Am. 1998;80:636–647.
Kondo T, Hirohashi S. Application of highly sensitive fluorescent dyes (CyDye DIGE Fluor saturation dyes) to laser microdissection and two-dimensional difference gel electrophoresis (2D-DIGE) for cancer proteomics. Nat Protoc. 2007;1:2940–2956.
Kondo T, Seike M, Mori Y, Fujii K, Yamada T, Hirohashi S. Application of sensitive fluorescent dyes in linkage of laser microdissection and two-dimensional gel electrophoresis as a cancer proteomic study tool. Proteomics. 2003;3:1758–1766.
Kusuzaki K, Takeshita H, Murata H, Hirata M, Hashiguchi S, Ashihara T, Hirasawa Y. Relation between cellular doxorubicin binding ability to nuclear DNA and histologic response to preoperative chemotherapy in patients with osteosarcoma. Cancer. 1998;82:2343–2349.
Ladanyi M. Fusions of the SYT and SSX genes in synovial sarcoma. Oncogene. 2001;5755–5762.
Lee YF, John M, Edwards S, Clark J, Flohr P, Maillard K, Edema M, Baker L, Mangham DC, Grimer R, Wooster R, Thomas JM, Fisher C, Judson I, Cooper CS. Molecular classification of synovial sarcomas, leiomyosarcomas and malignant fibrous histiocytomas by gene expression profiling. Br J Cancer. 2003;88:510–515.
Mariani L, Miceli R, Kattan MW, Brennan MF, Colecchia M, Fiore M, Casali PG, Gronchi A. Validation and adaptation of a nomogram for predicting the survival of patients with extremity soft tissue sarcoma using a three-grade system. Cancer. 2005;103:402–408.
Marina N, Gebhardt M, Teot L, Gorlick R. Biology and therapeutic advances for pediatric osteosarcoma. The Oncologist. 2004;9:422–441.
Milano A, Apice G, Ferrari E, Fazioli F, de Rosa V, de Luna AS, Iaffaioli RV, Caponigro F. New emerging drugs in soft tissue sarcoma. Crit Rev Oncol Hematol. 2006;59:74–84.
Mintz MB, Sowers R, Brown KM, Hilmer SC, Mazza B, Huvos AG, Meyers PA, LaFleur B, McDonough WS, Henry MM, Ramsey KE, Antonescu CR, Chen W, Healey JH, Daluski A, Berens ME, MacDonald TJ, Gorlick R, Stephan DA. An expression signature classifies chemotherapy-resistant pediatric osteosarcoma. Cancer Res. 2005;65:1748–1754.
Nagayama S, Katagiri T, Tsunoda T, Hosaka T, Nakashima Y, Araki N, Kusuzaki K, Nakayama T, Tsuboyama T, Nakamura T, Imamura M, Nakamura Y, Toguchida J. Genome-wide analysis of gene expression in synovial sarcomas using a cDNA microarray. Cancer Res. 2002;62:5859–5866.
Nakayama R, Nemoto T, Takahashi H, Ohta T, Kawai A, Seki K, Yoshida T, Toyama Y, Ichikawa H, Hasegawa T. Gene expression analysis of soft tissue sarcomas: characterization and reclassification of malignant fibrous histiocytoma. Mod Pathol. 2007;20:749–759.
Nielsen TO, West RB, Linn SC, Alter O, Knowling MA, O’Connell JX, Zhu S, Fero M, Sherlock G, Pollack JR, Brown PO, Botstein D, van de Rijn M. Molecular characterization of soft tissue tumours: a gene expression study. Lancet. 2002;359:1301–1307.
Oberlin O, Rey A, Desfachelles AS, Philip T, Plantaz D, Schmitt C, Plouvier E, Lejars O, Rubie H, Terrier P, Michon J. Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors. J Clin Oncol. 2006;24:3997–4002.
Ochi K, Daigo Y, Katagiri T, Nagayama S, Tsunoda T, Myoui A, Naka N, Araki N, Kudawara I, Ieguchi M, Toyama Y, Toguchida J, Yoshikawa H, Nakamura Y. Prediction of response to neoadjuvant chemotherapy for osteosarcoma by gene-expression profiles. Int J Oncol. 2004;24:647–655.
Resendes BL, Kuo SF, Robertson NG, Giersch ABS, Honrubia D, Ohara O, Adams JC, Morton CC. Isolation from cochlea of a novel human intronless gene with predominant fetal expression. J Assoc Res Otolaryngol. 2004;5:185–202.
Rosen G, Caparros B, Huvos AG, Kosloff C, Nirenberg A, Cacavio A, Marcove RC, Lane JM, Mehta B, Urban C. Preoperative chemotherapy for osteogenic sarcoma: selection of postoperative adjuvant chemotherapy based on the response of the primary tumor to preoperative chemotherapy. Cancer. 1982;15:1221–1230.
Suehara Y, Kondo T, Fujii K, Hasegawa T, Kawai A, Seki K, Beppu Y, Nishimura T, Kurosawa H, Hirohashi S. Proteomic signatures corresponding to histological classification and grading of soft-tissue sarcomas. Proteomics. 2006;6:4402–4409.
Suehara Y, Kondo T, Seki K, Shibata T, Fujii K, Goto M, Hasegawa T, Shimada Y, Sasako M, Shimoda T, Kurosawa H, Beppu Y, Kawai A, Hirohashi S. Pfetin, as a prognostic biomarker of gastrointestinal stromal tumors revealed by proteomics. Clin Cancer Res. 2008;14:1707–1717.
Tschoep K, Kohlmann A, Schlemmer M, Haferlach T, Issels RD. Gene expression profiling in sarcomas. Crit Rev Oncol Hematol. 2007;63:111–124.
Tunn PU, Schmidt-Peter P, Pomraenke D, Hohenberger P. Osteosarcoma in children: long-term functional analysis. Clin Orthop Relat Res. 2004;421:212–217.
Van der Zwan SM, DeMatteo RP. Gastrointestinal stromal tumor: 5 years later. Cancer. 2005;104:1781–1788.
Acknowledgments
We thank Robert Nakayama, Eisuke Kobayashi, and Ako Hosono for helpful and stimulating discussions and Misuzu Mori for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by a grant from the Ministry of Health, Labor and Welfare (AK, TK), a grant from the Ministry of Education, Science, Sports, and Culture (AK), and a grant from the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation of Japan (TK).
Each author certifies that his or her institution has approved the human protocol for this investigation and that all investigations were conducted in conformity with ethical principles of research.
About this article
Cite this article
Kawai, A., Kondo, T., Suehara, Y. et al. Global Protein-expression Analysis of Bone and Soft Tissue Sarcomas. Clin Orthop Relat Res 466, 2099–2106 (2008). https://doi.org/10.1007/s11999-008-0330-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11999-008-0330-4