
Abstract
Purpose of the Review
Kawasaki disease (KD) is a childhood systemic vasculitis of unknown etiology that causes coronary artery aneurysms (CAA), and if left undiagnosed can result in long-term cardiovascular complications and adult cardiac disease. Up to 20% of KD children fail to respond to IVIG, the mainstay of therapy, highlighting the need for novel therapeutic strategies. Here we review the latest findings in the field regarding specific etiology, genetic associations, and advancements in treatment strategies to prevent coronary aneurysms.
Recent Findings
Recent discoveries using the Lactobacillus casei cell wall extract (LCWE)-induced KD vasculitis mouse model have accelerated the study of KD pathophysiology and have advanced treatment strategies including clinical trials for IL-1R antagonist, Anakinra.
Summary
KD remains an elusive pediatric vasculitis syndrome and is the leading cause of acquired heart disease among children in the USA and developed countries. Advancements in combination treatment for refractory KD with further understanding of novel genetic risk factors serve as a solid foundation for future research endeavors in the field.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dr. Tomisaku Kawasaki, a Japanese pediatrician, was the first to publish a report of 50 patients with a peculiar constellation of symptoms including fever, several specific clinical findings, and cardiac complications in 1967 [1]. He received skepticism from other pediatricians at the time, who suggested that this Kawasaki disease (KD) was a self-limiting illness with no sequelae. Their tune quickly changed after 1970 when the first Japanese national survey of KD documented 10 autopsy cases of sudden cardiac death after KD [2]. By 1974, the link between KD and coronary artery vasculitis development was well established and accepted [3]. In this review, we will survey the current knowledge in KD pathophysiology, epidemiology, genetics, and advancement in treatment therapeutics.
KD Pathophysiology
KD is an acute febrile illness and systemic vasculitis of unknown etiology that predominantly afflicts children less than 5 years of age [4,5,6,7,8,9]. It often causes acute coronary as well as systemic arteritis, with coronary artery aneurysms (CAA) occurring in up to 30% of untreated patients, and can lead to ischemic heart disease, myocardial infarction, and even death, making it the leading cause of acquired heart disease in the USA [2, 10,11,12]. Once considered an acute self-limiting disease, KD now known to result in long-term complications of ongoing vascular remodeling and myocardial fibrosis [13]. While intravenous IgG (IVIG) treatment within the first 10 days of illness resolves inflammation and reduces the occurrence of coronary abnormalities like CAA from 25–30% down to 5–7% [14,15,16], up to a quarter of KD patients are IVIG-resistant and at higher risk for developing CAA [14]. Therefore, discovery of more effective treatments for KD is one of the highest priorities in pediatric research [17]. Coronary artery abnormalities in KD children are characterized histologically by inflammatory cell infiltration and focal destruction of the arterial media, especially elastic tissue in the media, with resultant CAA formation. Subsequent thrombosis or, less commonly, rupture of diseased coronary and other systemic vessels may occasionally be fatal. KD not only causes vessel inflammation in small and medium size arteries, but is also associated with myocarditis [18,19,20,21,22], which has been significantly under-investigated. Biopsy studies have documented that almost all KD patients develop myocarditis, which is associated with fatal arrhythmias and may lead to fibrosis [2, 10,11,12,13, 19, 23]. One study found that myocarditis was responsible for 13.6% of the total deaths in KD patients [24]. KD vasculitis is also now recognized to induce long-term vascular changes and remodeling such as luminal myofibroblast proliferation (LMP), leading to coronary artery stenosis with both cardiovascular and myocardial complications [13].
Epidemiology
KD has occurs globally; however, several ethnicities are noted to have a higher incidence [25••]. In North America, KD incidence is approximately 17 per 100,000 children, with the highest rates within the Pacific Islander and Asian populations at 30 per 100,000 [10] and lowest rates within the Caucasian populations at 9 per 100,000 children [26]. In Europe, the rate of KD is 5–10 per 100,000 in children under 5 years of age [27]. Higher prevalence of KD development has been reported in Asian countries, notably Japan, Korea, China, and Taiwan. In Japan, data from 2012 suggests incidence rates of 264.8 per 100,000 children younger than 5 years [28]. Higher rates in these populations, and in Asian children who live in the USA, suggest a genetic susceptibility that predicts a particular host response to the etiologic agent [29]. With regard to seasonality, there seems to be a winter-spring uptake in incidence of KD [10].
KD also tends to affect younger children, particularly those ≤ 5 years of age with a median age of onset around 9–11 months of life. Current thought suggests that passive maternal antibodies, which wane around 6–9 months after birth, may be protective, explaining why KD typically does not develop in children less than 3 months of age [27]. With regard to sex, KD is notably more common in males than females by a ratio of 1.5 to 1 [30]. Lastly, since no yearly fluctuations in KD incidence are observed, it is likely that the KD etiological triggering agent(s) is transmitted among household contacts and only triggers disease in genetically susceptible hosts [31].
Etiology and Pathophysiology
The etiology of KD remains unknown despite 40 years of intensive study. Several environmental factors that have been explored with regard to the etiology, including fine particulate air pollution and wind currents, although there is no clear evidence to show these factors affect disease pathogenesis [32, 33]. Indeed, it seems unlikely that environmental factors could be the sole reason for such a pervasive disease that occurs at all times of the year in virtually every country on the planet. More likely, KD is triggered by an infectious agent, although it is currently unidentified. Several overlaps exist between KD presentation and other infectious diseases, such as streptococcal and staphylococcal infections, and viral infections like adenovirus, measles, and glandular fever caused by Epstein-Barr virus (EBV) [34]. An infectious etiology is also evidenced by the seasonal prevalence of KD, which peaks in incidence during the winter and spring throughout many geographical areas [35]. Particularly in Japan, there is a notable, consistent uptick in cases with a bimodal peak in January and June/July, with a nadir in October [36]. In Japan, siblings of KD children are at increased risk for developing the disease [37]. Current dogma is that KD is triggered by an infectious agent entering through the mucosal surfaces, specifically the respiratory tract. The presence of oligoclonal IgA plasma cells in the inflamed tissues and coronary artery of KD patients, and the identification of an antigen-driven IgA response directed at intracytoplasmic inclusion bodies within the ciliated bronchial epithelium of KD patients, led to the hypothesis that KD infectious etiologic agent may be a RNA virus [38,39,40]. Recently, a protein with epitopes similar to hepacivirus C was detected in the intracytoplasmic inclusion bodies of KD patients, suggesting that a newer human virus similar to the single-stranded RNA hepaciviruses entering through the respiratory tract could be etiologically linked to KD [41].
Genetics
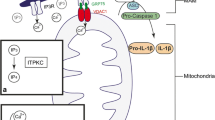
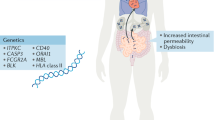
There are several risk factors that point toward heritability of risk for KD. In particular, increased KD incidence has been reported in first degree relatives [42]. Monozygotic twins have a 13% risk factor of developing KD when one of the twins acquires disease [43, 44]. Furthermore, retrospective reviews show that 1% of children admitted with KD have a family history of KD [45]. Single nucleotide polymorphisms (SNPs) in multiple genes, such as ITPKC [46], CASP3 [47, 48], FCGR2A [49], BLK [8], and CD40 [50] are associated with increased susceptibility for KD and CAA development, as comprehensively discussed in a recent review [51•]. The key SNPs are summarized in Table 1. Additionally, interleukin 1β (IL-1), a cytokine linked to the development of many inflammatory diseases [52], is implicated in human KD pathogenesis [53,54,55]. ITPKC acts upstream of IL-1 and NLRP3 inflammasome activation, and increased expression of ITPKC protein is associated with higher levels of intracellular calcium and increased production of IL-1β and IL-18 [56].
Clinical and Laboratory Diagnosis
Unlike many other diseases, diagnosis of KD is made based on the observation of clinical findings alone. An early diagnosis of KD is key for adequate treatment; however in the absence of a specific detection test, KD diagnosis is often overlooked and delayed. Diagnosis of complete or classic KD is based on a persistent fever lasting more than 5 days combined with four of the five major clinical criteria: (1) polymorphous rash or exanthem, (2) changes in the mucous membranes including cracked lips and strawberry tongue, (3) changes in the extremities (increased swelling of hands/feet and desquamation in the subacute phase), (4) cervical lymphadenopathy (typically unilateral), and (5) non-purulent conjunctival injection bilaterally with limbic sparing [28]. With regard to an “incomplete” KD diagnosis, patients must demonstrate ≤ 3 clinical criteria in the presence of fever for ≥ 5 days. Diagnostic KD criteria are shown in Table 2. The American Heart Association (AHA) issued new recommendations on diagnosis, treatment, and long-term management guidelines of KD, as well as an algorithm for incomplete KD diagnosis in 2017, including presence of CA abnormalities on echocardiogram and/or several laboratory abnormalities [28]. European consensus-based recommendations for the diagnosis and treatment of KD vasculitis (the SHARE initiative) were also recently published [57••]. It is important to highlight that KD symptoms often present one-by-one over time, rather than simultaneously. This combined with clinical findings that are common for other infectious febrile syndromes in childhood makes KD an often difficult diagnosis when presented to an inexperienced physician.
Prognostic Factors
Besides epidemiological and ethnic risk factors, it is well established that developing KD at a younger age (< 1 year) results in higher severity of coronary artery dilation compared with older children. One retrospective study showed that in infants < 6 months of age, there is up to a 68% incidence of coronary aneurysms [58]. It is also notable that children younger than 6 months have a higher prevalence of incomplete KD, which poses even more of a diagnostic challenge for clinicians as there is often delayed treatment, which further worsens the risk of CAA [59]. Even in young infants < 6 months of age where timely diagnosis and IVIG administration within the first 10 days of symptom onset is accomplished, nearly 20% had an aneurysm or giant aneurysm compared to 5% of cases that were in the ≥ 6-month age group [59]. Furthermore, serial echocardiogram monitoring is of utmost importance in this age group specifically, as an alarming 25% of infants < 6 months with an initial normal echocardiogram do end up developing dilated or aneurysmal coronary artery, usually within 2 weeks of treatment [59].
The natural history of CAA after KD diagnosis and treatment with IVIG is not well established. Many patients have regression of CAA to normal internal lumen diameter secondary to LMP and layering of thrombus in larger CAA [60]. In fact, in a study that followed KD patients for more than 30 years, CAA regression occurred in 75% of patients [61]. However, even in those in whom the lumen diameter returned to normal, the vascular wall may still be damaged, as there can be impaired dilation when the patient experiences increased cardiovascular demand [62].
Cardiac Complications and Long-Term Follow-up
KD is the leading cause of acquired heart disease among children in the USA and developed countries. KD-related cardiac complications can lead to significant morbidity and mortality. These only encompass CAA but can also include issues with myocardial contractility and in turn heart failure as well as other complications such as myocardial infarction, arrhythmias, and peripheral arterial occlusion. Although CAA receives most of the attention with regard to cardiac complications of KD, KD-related myocarditis is far more common from a histological perspective [23]. The majority of KD patients in the acute phase exhibit sub-clinical myocarditis, which presents with subtle electrocardiographic changes or mild clinical symptoms. In one clinical series, it was noted that an audible gallop was auscultated in 13% of patients within the first 20 days of illness. Other common clinical findings include tachycardia out of proportion to fever, hyperdynamic precordium, and gallop rhythm [63]. Though resolution of myocarditis after acute phase typically occurs, the long-term effects of the initial insult have been implicated in some of the long-term myocardial pathology, including fibrosis and myocyte drop. These changes that are notably independent of coronary artery abnormalities are still not fully delineated [28]. Several studies have also suggested that in a small subset of KD patients, diffuse myocarditis followed by myocardial fibrosis may lead to long-term systolic or diastolic dysfunction. This complication requires additional study, because most focus of previous research has been placed on coronary artery aneurysm complications [64].
Adult Complications of Childhood KD and Long-Term Follow-up
If no new therapeutic interventions are established, the estimated number of young adults with history of KD and coronary artery abnormalities is expected to grow by 1400 individuals per year [18]. Recently, a study looking at KD prevalence found that > 5% of all young adults (< 40 years of age) who undergo cardiac catheterization for suspected myocardial ischemia have aneurysms consistent with prior KD [65]. It is becoming increasingly important that adult cardiologists be comfortable and familiar with the signs and symptoms of acute KD to allow proper discussion with patients or parents regarding a previous KD-compatible illness that was not properly diagnosed. The complex inflammatory response associated with KD has the potential to affect multiple components of the cardiovascular system. In particular, vascular lesions observed in atherosclerosis differ from the coronary artery aneurysms that could progress to calcification and stenosis in the future [18]. Furthermore, in addition to causing aneurysms and stenoses, KD has been shown to create damaging effects on coronary artery function years after the acute presentation. Alarmingly, studies have demonstrated abnormal coronary flow reserve in patients with transiently dilated coronary arteries as well as in patients with evidence of inducible ischemia but normal-appearing coronary arteries via angiography [66]. The damage to the coronary arteries therefore likely extends beyond the scope of what is observed via angiography. In addition, cardiomyocyte drop and diffuse fibrosis noted in areas outside the watershed distribution of the epicardial coronary arteries have been documented in autopsies of adults late after KD [67]. Many deaths have also been noted secondary to presumptive ventricular arrhythmias secondary to left ventricular dysfunction in adults from Japan with history of prior KD [68]. Given these complications, adults with history of antecedent KD with regressed aneurysms should be followed regularly with functional and structural cardiovascular studies to delineate need for future interventions [18]. More systemic studies following adults with history of KD in childhood is necessary to further explore the natural history of this complex illness.
Experimental Mouse Model of KD and Acceleration of Discovery
While IVIG reduces the rate of CAA, morbidity and mortality associated with KD, lack of a specific etiologic agent, and incomplete understanding of the molecular mechanisms mediating KD cardiovascular pathology have hampered development of targeted and more effective treatment options. In addition, limited availability of human tissue samples has significantly impeded progress in our understanding of the etiology and pathology of KD, making the availability of a relevant animal model extremely valuable.
The well-described and well-accepted Lactobacillus casei cell wall extract (LCWE)-induced murine model of KD vasculitis and coronary arteritis closely mimics the important histological as well as immune-pathological features of the cardiovascular lesions (i.e., coronary arteritis, aortitis, myocarditis, aneurysms, including abdominal aorta aneurysms (AAA) seen in human KD) [69,70,71,72]. A single intraperitoneal injection of cell wall extract from LCWE reproducibly induces aortitis and proximal coronary arteritis (including epicardial coronary artery) that are histopathologically very similar to the coronary arteritis observed in human KD [69]. This KD murine model also allows the study of KD myocarditis as well as systemic arterial aneurysm formation, areas that have been neglected [73]. As seen in clinical KD, the LCWE-induced murine model of KD vasculitis is associated with systemic inflammation, increased body temperature, and pyrogens such as IL-1β and PGE2 [74, 75]. This mouse model also predicts therapeutic efficacy in children with KD [70, 72]. Indeed, currently used treatments in humans such as IVIG and anti-TNFα Ab were first shown to be beneficial in preventing CAA in the LCWE-induced KD vasculitis mouse model [70, 72]. This LCWE-induced murine model of KD vasculitis is highly dependent on IL-1β signaling and activation of the NLRP3 inflammasome, and treatment with the IL-1β antagonist Anakinra has beneficial effects [76,77,78], prompting clinical trials to test Anakinra in IVIG-resistant KD patients (NCT02179853) [79].
Treatment
Current expert consensus from the AHA recommends initial therapy with intravenous immune globulin (IVIG) at 2 g/kg administered over 10–12 h as soon as the diagnosis of KD can be made or within 10 days of presentation [28]. The mechanism of action of high dose IVIG is not fully understood, but is thought to be related to neutralization of the etiological agent and overall reduction of cytokine production [27, 80]. It should be given concurrently with moderate to high-dose (30–100 mg/kg/day) aspirin until the patient is afebrile [28]. Once the acute stage has resolved and the patient becomes afebrile and other inflammatory signs have resolved, it is prudent to transition to low-dose aspirin (3–5 mg/kg/day), until a repeat echocardiogram at 2 and 6–8 weeks after illness are documented within normal limits. There is always concern that patients may develop Reye syndrome while on low-dose aspirin; however, this risk is very low. In patients who develop influenza or varicella during low-dose aspirin course, it is recommended to transition to clopidogrel (Plavix®) briefly [60].
Of note, a sizable proportion (10–20%) of patients with KD can have an IVIG-refractory course [14, 81,82,83]. This subset is also more prone to developing CAA. Because of this, identifying patients who are at high risk of refractory course may be a beneficial treatment strategy. Multiple risk scores system, such as Kobayashi, Egami, and Sano scoring systems, has been developed based on clinical, laboratory, and demographic data obtained from Japanese KD patients in the goal to predict risk of IVIG resistance [84]. The Egami score, which takes into account the age of the patient, the number of days ill, and the levels of CRP and alanine aminotransferase to design a cutoff score of ≥ 3 for high risk of treatment resistance, has a 72% sensitivity and 76% specificity for IVIG resistance prediction in Japanese KD patients [85]. However, the Egami risk score has been shown unreliable for a multiethnic American population [14]. With the Kobayashi system, KD patients are classified into either low or high risk of developing IVIG resistance based on levels of sodium, aspartate aminotransferase, CRP, percentages of neutrophils, platelet counts, and patient age [86]. We know now that in non-Japanese children, a coronary artery Z-score of ≥ 3 or age < 6 months will have higher initial risk of CAA. In addition, a Kobayashi score of ≥ 5 in Japanese children indicates higher risk for development of IVIG-failure and eventual CAA development and may warrant more aggressive initial treatment plan [86]. There lies some controversy in use of Kobayashi score in non-Asian populations. One retrospective population-based cohort study in Germany assessed the prognostic validity of Kobayashi and Egami scoring systems for refractory KD, and found that while the relative risk for those with positive scores ranged between 2.32 and 3.73, the prognostic properties were low (likelihood ratio positive: 1.83–4.57; sensitivity range of 0.28–0.53) [87]. None of these scoring systems proved to be appropriate predictors of CAA 1 year after active illness in high-risk Caucasian children with KD [87].
IVIG-Non-response and Use of Anti-TNFα Monoclonal Antibody (Infliximab)
For patients who do develop true refractory KD, defined as resurgence of fever (> 100.4 °F) within 36–72 h after IVIG completion, the current recommendation is to give a second dose of IVIG at the same concentration and rate. Alternatively, Infliximab, an anti-TNFα monoclonal antibody, can be used in place of the second dose of IVIG, as in a retrospective trial a single dose of IV Infliximab given at 5 mg/kg IV over 2 h showed improvement in fever curve as well as decreased hospital days, but longer term coronary artery outcomes and adverse events were similar to the round two IVIG group [88]. TNFα is a pro-inflammatory cytokine that is elevated in the acute phase of illness. Infliximab has also been explored as a primary therapeutic alternative in conjunction with IVIG. In one study, combination of these interventions resulted in fewer days of fever, rapid drop in CRP, and faster reduction of CA Z-score of LAD compared to IVIG alone within the first 2 weeks of administration [89]. However, at the 5-week mark, none of the laboratory values differed significantly between the groups compared to baseline. Though the addition of infliximab to IVIG as primary treatment did not decrease the IVIG resistance, it did prove to be safe and well-tolerated in children less than 1 year of age [89]. The KIDCARE trial (The Kawasaki Disease Comparative Effectiveness Trial) is a 30 site, phase 3, randomized trial for a second dose of IVIG versus infliximab for resistant KD with the caveat that a higher dose of 10 mg/kg infusion will be studied. This dosing adjustment would be double that of prior studies [90].
The Role of Corticosteroids
Corticosteroid was initially used as mainstay of KD treatment before efficacy of IVIG was established [91]. However, a randomized control trial for treatment for acute KD with intravenous methylprednisolone versus a placebo prior to conventional therapy with IVIG did not support addition of pulsed IV steroids, and showed no difference among groups regarding change in CAA Z-score, number of hospital days, or adverse events [82]. The current paradigm supports use of corticosteroids in patients who are at high risk of KD-related complications after conventional IVIG is given. Dosing is usually prednisolone 2 mg/kg/day IV divided every 8 h until afebrile, sometimes with a tapered steroid regimen to follow in addition to a second round of IVIG [28]. An open-label, randomized, blinded endpoint study (the RAISE study) where KD Japanese patients predicted as IVIG non-responder with the Kobayashi score were treated with prednisolone and standard IVIG therapy for 4–5 weeks or until normalized C-reactive protein levels demonstrated that IVIG and prednisolone combinational treatment decreased days of fever and lead to a greater reduction in coronary artery Z-scores [92]. Importantly, scoring systems used to predict IVIG resistance risk are highly sensitive and specific for the Japanese population, yet they perform poorly in a multiethnic North American population [14], which may be one limitation of the RAISE study.
Adjunctive Therapies
Anakinra
The pathogenic role of IL-1β pathway has been well-demonstrated in the LCWE murine model of KD [76,77,78]. Recently, work in this model linked elevated IL-1β serum concentrations with increased intestinal permeability and mucosal barrier dysfunction [93]. Those findings suggest that IL-1β activation lies upstream of disrupted intestinal barrier function, leading to subsequent IgA vasculitis and cardiac inflammation [93]. This provides the background for a possible future role of treatments targeting the IL-1β pathway, including Anakinra, an IL-1 receptor antagonist. Upregulation of the IL-1 pathway in KD has been well established [53, 56], including validation of key genes in the IL-1 pathway in an independent cohort of 20 KD patients and 10 healthy controls [94].
Anakinra competitively inhibits both IL-1α and IL-1β from binding to IL-1 type 1 receptor, with rapid onset with a safe profile in infants and children [95]. It is currently utilized in several inflammatory conditions in infants and children including systemic juvenile idiopathic arthritis as well as other less common autoinflammatory conditions such as cryopyrin-associated periodic syndromes (CAPS) and tumor necrosis factor receptor-associated periodic syndrome (TRAPS) [96,97,98]. Anakinra has also been used to treat severe, refractory KD in patients with treatment failure to IVIG and steroids [99]. Most recently, several case reports using Anakinra have successfully treated complicated cases in patients with refractory KD [100,101,102,103] as well as in macrophage activation syndrome caused by refractory KD [104].
A study to determine the safety, pharmacokinetics, and activity of Anakinra in acute KD patients is currently underway in two national study sites. The phase I/IIa trial, ANAKID, will evaluate children greater than 8 months of age with a CA Z-score of at least ≥ 3.0 in the right coronary artery (RCA) and/or left anterior descending (LAD) artery. This dose escalation study will monitor primary outcome measures of Anakinra safety at a 2- or 6-week course [79]. Another phase II clinical study investigating the role of Anakinra in IVIG-resistant KD patients has been successfully conducted in France, and the results have been submitted for publication (Personal communication from Isabelle Kone-Paut).
Etanercept
While infliximab, a monoclonal antibody against TNFα, has no impact on IVIG resistance, a similar but slightly different drug, etanercept, has recently been evaluated for its role in KD [105]. Etanercept is a soluble TNF receptor fusion protein that antagonizes endogenous TNF. It is administered subcutaneously weekly, compared to infliximab which can be given less frequently. One potential disadvantage of infliximab is the possibility of antidrug antibodies developing during treatment; however, etanercept is less immunogenic and does not have this problem [106]. Still, a recent phase 3, multicenter, placebo-controlled, double blind randomized trial, EATAK (Etanercept as Adjunctive Treatment for Acute Kawasaki Disease), found no significant benefit to use of etanercept in IVIG resistance [105]. In patients that were > 1 year of age however, there was some benefit. Etanercept was well tolerated however and showed no difference in safety in comparison to placebo. Future studies further stratifying patients by high-risk genotypes or demographics would be valuable with regard to clinical implementation of this therapeutic option [105].
Cyclosporine
Cyclosporine is in theory an ideal drug to prevent progression of inflammation in the arterial wall given that it is a specific T cell inhibitor that blocks the calcium-driven calcineurin-NFAT pathway leading to the transcription and release of key pathogenic pro-inflammatory cytokines that are pivotal for KD development [107]. Studies have shown that CD8+T cells infiltrate the arterial wall after initial arrival of neutrophils and macrophages in KD CAA [108, 109]. The use of cyclosporine combined with IVIG in refractory KD patients was examined in a recently published Japanese phase III randomized control trial [110]. Cyclosporine was overall well tolerated and showed improvement in CAA at the 4-week mark in patients with a coronary artery Z-sore of greater than 3.0 or larger more efficiently than conventional treatment with IVIG and high-dose aspirin [110]. These findings however must be taken with caution, as the risk scoring system used in this trial had adequate specificity (87%) but poor sensitivity (33%) for prediction of IVIG non-responders in European populations [110]. In a recent comment on this study, it was proposed that a better design for a similar clinical trial for non-Japanese populations should enroll patients with initial coronary artery Z-sores of > 2.5, as about 80% of patients that progress to develop CAAs have a Z-score above this threshold on initial electrocardiogram [111]. Additional studies should be conducted to determine which specific populations of KD patients would benefit most from this combination therapy as initial treatment.
Atorvastatin
Most recently, in search of novel treatments for CAA inflammation in KD patients, research has turned to exploring anti-inflammatory and antioxidant effects of statin drugs. HMG-co-A reductase inhibitors are notable for promoting healing of the endothelium with significant risk reduction in adults with cardiovascular risks [112]. Statin drugs have been shown to induce autophagy and mitophagy, thus stopping NLRP3 inflammasome pathway from activating additional inflammatory cytokines like IL-1β which is closely linked to KD pathogenesis [113]. Recently a new phase I/IIa study to measure the safety and tolerability in patients with KD after having received standard IVIG, aspirin, and infliximab prior to study entry was completed [114]. This was the first dose-escalation pharmacokinetic (PK) study with statin use in children with acute cardiovascular inflammation and the first study to demonstrate the safety of atorvastatin (FDA approved for children ≥ 8 years of age). Though the sample size was smaller, and the age range of patients only included children with KD and CAA involvement greater than age 2, it demonstrated that atorvastatin was safe and well tolerated. Future phase III efficacy trials should be conducted to further delineate the immunomodulatory and anti-inflammatory effects of statins in children with KD [115]. This two-center study was based partially on the rationale that IVIG reduces inflammation by increasing expression of IL-10 and thus increasing regulatory T cells. Atorvastatin has been shown to also increase the pool of TREG cells in adults that have rheumatoid arthritis, thus reducing disease [116].
Timing of Immunizations After IVIG in Pediatric Populations
After standard treatment of IVIG in KD pediatric patients, it is important to note that there is ongoing concern of the use of high dose IVIG and its interference of serological responses to active immunization [117]. Current guidelines from the Advisory Committee on Immunization Practices and the American Academy of Pediatrics advise giving the measles vaccine after an interval of 11 months or more post-IVIG. This is based on data from a study which showed that an intramuscular dose of immunoglobulin at 80 mg/kg, much less than that recommended for KD, inhibited serological response to measles for up to 5 months [117]. Though this is recommended, there is still no clear consensus regarding the appropriate time interval. In Japan for example, common practice is to give MMR vaccine at least 6–7 months after administration of IVIG. This is based on a study that also measured similar antibodies of measles from IVIG [118]. In one recent retrospective study, evaluating geometric mean concentrations (GMC) in 58 patients vaccinated after IVIG administration, patients had reduced effectiveness of mumps and rubella vaccination for up to 6 months, and this was extended to 9 months for measles. Therefore, vaccinations, particularly the MMR vaccine, should be deferred at least 9 months after IVIG administration and there should be more awareness for these post-KD issues [119].
With regard to inactivated vaccines and live oral vaccines (rotavirus, typhoid, and polio, in countries where it is still administrated, live intranasal vaccine, BCG vaccine, and yellow fever vaccine), all of these can be administered at any time after high dose IVIG administration in treatment of KD. Of note, inactivated influenza vaccine should be administered to all patients with history of KD on aspirin therapy given concerns and risk of Reye syndrome if the patient develops infection with influenza. There is a small theoretical risk of Reye syndrome being induced by the attenuated VZ virus of the vaccine; therefore, this vaccine should be given at least 2 days after temporary stop of aspirin dosing, which should then be replaced by clopidogrel (Plavix) for 6 weeks after varicella vaccine administration [120].
Conclusions
At present, our complete understanding of KD is limited by our lack of an identified etiologic agent. However, many advancements have been made over the decades with regard to understanding of natural course of this entity with the aid of mouse models and human studies alike. Many studies have aided in our understanding of management of complicated cases of KD, and several therapeutic options are becoming more widely available for use in high-risk patients. As we enter a new decade of research, many of the children with previous KD are entering adulthood, and long-term follow-up to address possible cardiac complications is now becoming increasingly relevant for practitioners worldwide.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Arerugi = [Allergy]. 1967;16(3):178–222.
Burns JC. Kawasaki disease update. Indian J Pediatr. 2009;76(1):71–6.
Burns JC, Kushner HI, Bastian JF, Shike H, Shimizu C, Matsubara T, et al. Kawasaki disease: a brief history. Pediatrics. 2000;106(2):E27.
Kawasaki T, Kosaki F, Okawa S, Shigematsu I, Yanagawa H. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics. 1974;54(3):271–6.
Rowley AH, Baker SC, Orenstein JM, Shulman ST. Searching for the cause of Kawasaki disease—cytoplasmic inclusion bodies provide new insight. Nat Rev Microbiol. 2008;6(5):394–401.
Rowley AH, Baker SC, Shulman ST, Garcia FL, Fox LM, Kos IM, et al. RNA-containing cytoplasmic inclusion bodies in ciliated bronchial epithelium months to years after acute Kawasaki disease. PLoS One. 2008;3(2):e1582.
Rowley AH, Shulman ST, Garcia FL, Guzman-Cottrill JA, Miura M, Lee HL, et al. Cloning the arterial IgA antibody response during acute Kawasaki disease. J Immunol. 2005;175(12):8386–91.
Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet. 2012;44(5):517–21.
Shimizu C, Matsubara T, Onouchi Y, Jain S, Sun S, Nievergelt CM, et al. Matrix metalloproteinase haplotypes associated with coronary artery aneurysm formation in patients with Kawasaki disease. J Hum Genet. 2010;55(12):779–84.
Holman R, Belay E, Christensen K, Folkema A, Steiner C, Schonberger L. Hospitalizations for Kawasaki syndrome among children in the United States, 1997-2007. Pediatr Infect Dis J. 2010;29(6):483–8.
Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young American Heart Association. Circulation. 2004;110(17):2747–71.
Burns JC, Glodé MP. Kawasaki syndrome. Lancet. 2004;364(9433):533–44.
Orenstein JM, Shulman ST, Fox LM, Baker SC, Takahashi M, Bhatti TR, et al. Three linked vasculopathic processes characterize Kawasaki disease: a light and transmission electron microscopic study. PLoS One. 2012;7(6):e38998.
Tremoulet AH, Best BM, Song S, Wang S, Corinaldesi E, Eichenfield JR, et al. Resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatr. 2008;153(1):117–21.
Sundel RP, Burns JC, Baker A, Beiser AS, Newburger JW. Gamma globulin re-treatment in Kawasaki disease. J Pediatr. 1993;123(4):657–9.
Burns JC, Capparelli EV, Brown JA, Newburger JW, Glode MP. Intravenous gamma-globulin treatment and retreatment in Kawasaki disease. US/Canadian Kawasaki Syndrome Study Group. Pediatr Infect Dis J. 1998;17(12):1144–8.
Rowley AH. Finding the cause of Kawasaki disease: a pediatric infectious diseases research priority. J Infect Dis. 2006;194(12):1635–7.
Gordon JB, Kahn AM, Burns JC. When children with Kawasaki disease grow up: myocardial and vascular complications in adulthood. J Am Coll Cardiol. 2009;54(21):1911–20.
Yutani C, Okano K, Kamiya T, Oguchi K, Kozuka T, Ota M, et al. Histopathological study on right endomyocardial biopsy of Kawasaki disease. Br Heart J. 1980;43(5):589–92.
Yonesaka S, Takahashi T, Matubara T, Nakada T, Furukawa H, Tomimoto K, et al. Histopathological study on Kawasaki disease with special reference to the relation between the myocardial sequelae and regional wall motion abnormalities of the left ventricle. Jpn Circ J. 1992;56(4):352–8.
Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114(6):1708–33.
Takahashi M. Myocarditis in Kawasaki syndrome. A minor villain? Circulation. 1989;79(6):1398–400.
Dahdah N. Not just coronary arteritis, Kawasaki disease is a myocarditis, too. J Am Coll Cardiol. 2010;55(14):1507.
Ayusawa M, Abe O, Miyashita T. The study of death cases in acute phase Kawasaki disease over the last 10 years based on a National Survey [in Japanese]. Pediatr Cardiol Cardiovasc Surg. 2000;20:245.
•• McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation. 2017;135(17):e927–e99 State of the art and most updated American Heart Association (AHA) evidence-based guidelines for Kawasaki Disease diagnostic, management and treatment.
Hedrich CM, Schnabel A, Hospach T. Kawasaki disease. Front Pediatr. 2018;6:198.
Dietz SM, van Stijn D, Burgner D, Levin M, Kuipers IM, Hutten BA, et al. Dissecting Kawasaki disease: a state-of-the-art review. Eur J Pediatr. 2017;176(8):995–1009.
McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135(17):e927–e99.
Rowley AH. Is Kawasaki disease an infectious disorder? Int J Rheum Dis. 2018;21(1):20–5.
Tacke CE, Breunis WB, Pereira RR, Breur JM, Kuipers IM, Kuijpers TW. Five years of Kawasaki disease in the Netherlands: a national surveillance study. Pediatr Infect Dis J. 2014;33(8):793–7.
Nagao Y, Urabe C, Nakamura H, Hatano N. Predicting the characteristics of the aetiological agent for Kawasaki disease from other paediatric infectious diseases in Japan. Epidemiol Infect. 2016;144(3):478–92.
Rodo X, Curcoll R, Robinson M, Ballester J, Burns JC, Cayan DR, et al. Tropospheric winds from northeastern China carry the etiologic agent of Kawasaki disease from its source to Japan. Proc Natl Acad Sci U S A. 2014;111(22):7952–7.
Rodo X, Ballester J, Curcoll R, Boyard-Micheau J, Borras S, Morgui JA. Revisiting the role of environmental and climate factors on the epidemiology of Kawasaki disease. Ann N Y Acad Sci. 2016;1382(1):84–98.
Harnden A, Takahashi M, Burgner D. Kawasaki disease. BMJ. 2009;338:b1514.
Uehara R, Belay ED. Epidemiology of Kawasaki disease in Asia, Europe, and the United States. J Epidemiol. 2012;22(2):79–85.
Burns JC, Cayan DR, Tong G, Bainto EV, Turner CL, Shike H, et al. Seasonality and temporal clustering of Kawasaki syndrome. Epidemiology. 2005;16(2):220–5.
Yanagawa H, Nakamura Y, Yashiro M, Fujita Y, Nagai M, Kawasaki T, et al. A nationwide incidence survey of Kawasaki disease in 1985-1986 in Japan. J Infect Dis. 1988;158(6):1296–301.
Rowley AH, Baker SC, Shulman ST, Rand KH, Tretiakova MS, Perlman EJ, et al. Ultrastructural, immunofluorescence, and RNA evidence support the hypothesis of a new virus associated with Kawasaki disease. J Infect Dis. 2011;203(7):1021–30.
Rowley AH, Shulman ST, Mask CA, Finn LS, Terai M, Baker SC, et al. IgA plasma cell infiltration of proximal respiratory tract, pancreas, kidney, and coronary artery in acute Kawasaki disease. J Infect Dis. 2000;182(4):1183–91.
Rowley AH, Shulman ST, Spike BT, Mask CA, Baker SC. Oligoclonal IgA response in the vascular wall in acute Kawasaki disease. J Immunol. 2001;166(2):1334–43.
Rowley AH, Gruen LJ, Bodnar T, Innocentini N, Shulman ST. Monoclonal antibodies from children with Kawasaki disease (KD) recognize hepacivirus peptides: Abstract-Pediatric Academic Society; 2019.
Dergun M, Kao A, Hauger SB, Newburger JW, Burns JC. Familial occurrence of Kawasaki syndrome in North America. Arch Pediatr Adolesc Med. 2005;159(9):876–81.
Fujita Y, Nakamura Y, Sakata K, Hara N, Kobayashi M, Nagai M, et al. Kawasaki disease in families. Pediatrics. 1989;84(4):666–9.
Kottek A, Shimizu C, Burns JC. Kawasaki disease in monozygotic twins. Pediatr Infect Dis J. 2011;30(12):1114–6.
Yanagawa H, Nakamura Y, Yashiro M, Ojima T, Koyanagi H, Kawasaki T. Update of the epidemiology of Kawasaki disease in Japan—from the results of 1993-94 nationwide survey. J Epidemiol. 1996;6(3):148–57.
Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. 2008;40(1):35–42.
Kuo HC, Hsu YW, Wu CM, Chen SH, Hung KS, Chang WP, et al. A replication study for association of ITPKC and CASP3 two-locus analysis in IVIG unresponsiveness and coronary artery lesion in Kawasaki disease. PLoS One. 2013;8(7):e69685.
Onouchi Y, Ozaki K, Buns JC, Shimizu C, Hamada H, Honda T, et al. Common variants in CASP3 confer susceptibility to Kawasaki disease. Hum Mol Genet. 2010;19(14):2898–906.
Khor CC, Davila S, Breunis WB, Lee YC, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. 2011;43(12):1241–6.
Lee YC, Kuo HC, Chang JS, Chang LY, Huang LM, Chen MR, et al. Two new susceptibility loci for Kawasaki disease identified through genome-wide association analysis. Nat Genet. 2012;44(5):522–5.
• Onouchi Y. The genetics of Kawasaki disease. Int J Rheum Dis. 2018;21(1):26–30 An important review that summarizes the key human genetic variants associated with increased Kawasaki Disease susceptibility.
Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6(4):232–41.
Leung DY, Cotran RS, Kurt-Jones E, Burns JC, Newburger JW, Pober JS. Endothelial cell activation and high interleukin-1 secretion in the pathogenesis of acute Kawasaki disease. Lancet. 1989;2(8675):1298–302.
Suzuki H, Uemura S, Tone S, Iizuka T, Koike M, Hirayama K, et al. Effects of immunoglobulin and gamma-interferon on the production of tumour necrosis factor-alpha and interleukin-1 beta by peripheral blood monocytes in the acute phase of Kawasaki disease. Eur J Pediatr. 1996;155(4):291–6.
Weng KP, Hsieh KS, Ho TY, Huang SH, Lai CR, Chiu YT et al. IL-1B polymorphism in association with initial intravenous immunoglobulin treatment failure in Taiwanese children with Kawasaki disease. Circ J. 2010;74(3):544–51.
Alphonse MP, Duong TT, Shumitzu C, Hoang TL, McCrindle BW, Franco A, et al. Inositol-triphosphate 3-kinase C mediates Inflammasome activation and treatment response in Kawasaki disease. J Immunol. 2016;197(9):3481–9.
•• de Graeff N, Groot N, Ozen S, Eleftheriou D, Avcin T, Bader-Meunier B, et al. European consensus-based recommendations for the diagnosis and treatment of Kawasaki disease—the SHARE initiative. Rheumatology (Oxford). 2019;58(4):672–82 State of the art and most updated Single Hub and Access point for pediatric Rheumatology in Europe (SHARE) evidence based guidelines for Kawasaki Disease diagnostic, management and treatment.
Cameron SA, Carr M, Pahl E, DeMarais N, Shulman ST, Rowley AH. Coronary artery aneurysms are more severe in infants than in older children with Kawasaki disease. Arch Dis Child. 2019;104(5):451–5.
Salgado AP, Ashouri N, Berry EK, Sun X, Jain S, Burns JC, et al. High risk of coronary artery aneurysms in infants younger than 6 months of age with Kawasaki disease. J Pediatr. 2017;185:112–6 e1.
Rowley AH, Shulman ST. Pathogenesis and management of Kawasaki disease. Expert Rev Anti-Infect Ther. 2010;8(2):197–203.
Friedman KG, Gauvreau K, Hamaoka-Okamoto A, Tang A, Berry E, Tremoulet AH et al. Coronary artery aneurysms in Kawasaki disease: risk factors for progressive disease and adverse cardiac events in the US population. J Am Heart Assoc. 2016;5(9).
Furuyama H, Odagawa Y, Katoh C, Iwado Y, Ito Y, Noriyasu K, et al. Altered myocardial flow reserve and endothelial function late after Kawasaki disease. J Pediatr. 2003;142(2):149–54.
Dionne A, Dahdah N. Myocarditis and Kawasaki disease. Int J Rheum Dis. 2017;21(1):45–9.
Nagasawa H, Arakaki Y, Yamada O, Nakajima T, Kamiya T. Longitudinal observations of left ventricular end-diastolic dimension in children using echocardiography. Pediatr Cardiol. 1996;17(3):169–74.
Daniels LB, Tjajadi MS, Walford HH, Jimenez-Fernandez S, Trofimenko V, Fick DB Jr, et al. Prevalence of Kawasaki disease in young adults with suspected myocardial ischemia. Circulation. 2012;125(20):2447–53.
Suzuki A, Yamagishi M, Kimura K, Sugiyama H, Arakaki Y, Kamiya T, et al. Functional behavior and morphology of the coronary artery wall in patients with Kawasaki disease assessed by intravascular ultrasound. J Am Coll Cardiol. 1996;27(2):291–6.
Rozin L, Koehler SA, Shakir A, Ladham S, Wecht CH. Kawasaki disease: a review of pathologic features of stage IV disease and two cases of sudden death among asymptotic young adults. Am J Forensic Med Pathol. 2003;24(1):45–50.
Tsuda E, Arakaki Y, Shimizu T, Sakaguchi H, Yoshimura S, Yazaki S, et al. Changes in causes of sudden deaths by decade in patients with coronary arterial lesions due to Kawasaki disease. Cardiol Young. 2005;15(5):481–8.
Lehman TJ, Walker SM, Mahnovski V, McCurdy D. Coronary arteritis in mice following the systemic injection of group B Lactobacillus casei cell walls in aqueous suspension. Arthritis Rheum. 1985;28(6):652–9.
Myones BL, Bathoria JM, Lehman TJ, Shulman ST, editors. Human IVIG inhibits Lactobacillus casei-inducible coronary arteritis in a murine model1995 1995/01/01: The 5th International Kawasaki Disease Symposium.
Schulte DJ, Yilmaz A, Shimada K, Fishbein MC, Lowe EL, Chen S, et al. Involvement of innate and adaptive immunity in a murine model of coronary arteritis mimicking Kawasaki disease. J Immunol. 2009;183(8):5311–8.
Lehman TJA, Sherry B, Gietl DM, Nguyen HT, Cerami A. Suppression of lactobacillus casei cell wall-induced coronary arteritis in mice by antibody to murine tumor necrosis factor. Proc Third Int Conf Kawasaki Disease. 1988:203–206.
Matundan HH, Sin J, Rivas MN, Fishbein MC, Lehman TJ, Chen S, et al. Myocardial fibrosis after adrenergic stimulation as a long-term sequela in a mouse model of Kawasaki disease vasculitis. JCI Insight. 2019;4(3).
Maury CP, Salo E, Pelkonen P. Circulating interleukin-1 beta in patients with Kawasaki disease. N Engl J Med. 1988;319(25):1670–1.
Lee T, Furukawa S, Fukuda Y, Yabuta K, Kato H. Plasma prostaglandin E 2 level in Kawasaki disease. Prostaglandins Leukot Essent Fat Acids. 1988;31(2):53–7.
Lee Y, Schulte DJ, Shimada K, Chen S, Crother TR, Chiba N, et al. Interleukin-1beta is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. 2012;125(12):1542–50.
Lee Y, Wakita D, Dagvadorj J, Shimada K, Chen S, Huang G, et al. IL-1 signaling is critically required in stromal cells in Kawasaki disease vasculitis mouse model: role of both IL-1alpha and IL-1beta. Arterioscler Thromb Vasc Biol. 2015;35(12):2605–16.
Wakita D, Kurashima Y, Crother TR, Noval Rivas M, Lee Y, Chen S, et al. Role of interleukin-1 signaling in a mouse model of Kawasaki disease-associated abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2016;36(5):886–97.
Tremoulet AH, Jain S, Kim S, Newburger J, Arditi M, Franco A, et al. Rationale and study design for a phase I/IIa trial of anakinra in children with Kawasaki disease and early coronary artery abnormalities (the ANAKID trial). Contemp Clin Trials. 2016;48:70–5.
Burns JC, Franco A. The immunomodulatory effects of intravenous immunoglobulin therapy in Kawasaki disease. Expert Rev Clin Immunol. 2015;11(7):819–25.
Moffett BS, Syblik D, Denfield S, Altman C, Tejtel-Sexson K. Epidemiology of immunoglobulin resistant Kawasaki disease: results from a large, national database. Pediatr Cardiol. 2015;36(2):374–8.
Newburger JW, Sleeper LA, McCrindle BW, Minich LL, Gersony W, Vetter VL, et al. Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease. N Engl J Med. 2007;356(7):663–75.
Bar-Meir M, Kalisky I, Schwartz A, Somekh E, Tasher D. Prediction of resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatric Infect Dis Soc. 2018;7(1):25–9.
Rigante D, Andreozzi L, Fastiggi M, Bracci B, Natale MF, Esposito S. Critical overview of the risk scoring systems to predict non-responsiveness to intravenous immunoglobulin in Kawasaki syndrome. Int J Mol Sci. 2016;17(3):278.
Egami K, Muta H, Ishii M, Suda K, Sugahara Y, Iemura M, et al. Prediction of resistance to intravenous immunoglobulin treatment in patients with Kawasaki disease. J Pediatr. 2006;149(2):237–40.
Kobayashi T, Inoue Y, Takeuchi K, Okada Y, Tamura K, Tomomasa T, et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation. 2006;113(22):2606–12.
Jakob A, von Kries R, Horstmann J, Hufnagel M, Stiller B, Berner R, et al. Failure to predict high-risk Kawasaki disease patients in a population-based study cohort in Germany. Pediatr Infect Dis J. 2018;37(9):850–5.
Son MB, Gauvreau K, Burns JC, Corinaldesi E, Tremoulet AH, Watson VE, et al. Infliximab for intravenous immunoglobulin resistance in Kawasaki disease: a retrospective study. J Pediatr. 2011;158(4):644–9 e1.
Tremoulet AH, Jain S, Jaggi P, Jimenez-Fernandez S, Pancheri JM, Sun X, et al. Infliximab for intensification of primary therapy for Kawasaki disease: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet. 2014;383(9930):1731–8.
Roberts SC, Jain S, Tremoulet AH, Kim KK, Burns JC, Anand V, et al. The Kawasaki Disease Comparative Effectiveness (KIDCARE) trial: a phase III, randomized trial of second intravenous immunoglobulin versus infliximab for resistant Kawasaki disease. Contemp Clin Trials. 2019;79:98–103.
Furusho K, Kamiya T, Nakano H, Kiyosawa N, Shinomiya K, Hayashidera T, et al. High-dose intravenous gammaglobulin for Kawasaki disease. Lancet. 1984;2(8411):1055–8.
Kobayashi T, Saji T, Otani T, Takeuchi K, Nakamura T, Arakawa H, et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. Lancet. 2012;379(9826):1613–20.
Noval Rivas M, Wakita D, Franklin MK, Carvalho TT, Abolhesn A, Gomez AC, et al. Intestinal permeability and IgA provoke immune vasculitis linked to cardiovascular inflammation. Immunity. 2019;51(3):508–21.e6.
Hoang LT, Shimizu C, Ling L, Naim ANM, Khor CC, Tremoulet AH, et al. Global gene expression profiling identifies new therapeutic targets in acute Kawasaki disease. Genome Med. 2014;6(11):541.
Sota J, Vitale A, Insalaco A, Sfriso P, Lopalco G, Emmi G, et al. Safety profile of the interleukin-1 inhibitors anakinra and canakinumab in real-life clinical practice: a nationwide multicenter retrospective observational study. Clin Rheumatol. 2018;37(8):2233–40.
Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355(6):581–92.
Ringold S, Weiss PF, Beukelman T, DeWitt EM, Ilowite NT, Kimura Y, et al. 2013 update of the 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: recommendations for the medical therapy of children with systemic juvenile idiopathic arthritis and tuberculosis screening among children receiving biologic medications. Arthritis Rheum. 2013;65(10):2499–512.
Hoffman HM. Therapy of autoinflammatory syndromes. J Allergy Clin Immunol. 2009;124(6):1129–38 quiz 39-40.
Cohen S, Tacke CE, Straver B, Meijer N, Kuipers IM, Kuijpers TW. A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygenation. Ann Rheum Dis. 2012;71(12):2059–61.
Blonz G, Lacroix S, Benbrik N, Warin-Fresse K, Masseau A, Trewick D et al. Severe late-onset Kawasaki disease successfully treated with Anakinra. J Clin Rheumatol. 2018:1. https://doi.org/10.1097/RHU.0000000000000814.
Guillaume MP, Reumaux H, Dubos F. Usefulness and safety of anakinra in refractory Kawasaki disease complicated by coronary artery aneurysm. Cardiol Young. 2018;28(5):739–42.
Shafferman A, Birmingham JD, Cron RQ. High dose Anakinra for treatment of severe neonatal Kawasaki disease: a case report. Pediatr Rheumatol Online J. 2014;12:26.
Sánchez-Manubens J, Gelman A, Franch N, Teodoro S, Palacios JR, Rudi N, et al. A child with resistant Kawasaki disease successfully treated with anakinra: a case report. BMC Pediatr. 2017;17(1):102.
Lind-Holst M, Hartling UB, Christensen AE. High-dose anakinra as treatment for macrophage activation syndrome caused by refractory Kawasaki disease in an infant. BMJ Case Rep. 2019;12(8).
Portman MA, Dahdah NS, Slee A, Olson AK, Choueiter NF, Soriano BD et al. Etanercept with IVIg for acute Kawasaki disease: a randomized controlled trial. Pediatrics. 2019;143(6):e20183675.
Vincent FB, Morand EF, Murphy K, Mackay F, Mariette X, Marcelli C. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)-specific neutralising agents in chronic inflammatory diseases: a real issue, a clinical perspective. Ann Rheum Dis. 2013;72(2):165–78.
Onouchi Y. Genetics of Kawasaki disease. Circ J. 2012;76(7):1581–6.
Brown TJ, Crawford SE, Cornwall ML, Garcia F, Shulman ST, Rowley AH. CD8 T lymphocytes and macrophages infiltrate coronary artery aneurysms in acute Kawasaki disease. J Infect Dis. 2001;184(7):940–3.
Rowley AH, Wylie KM, Kim KY, Pink AJ, Yang A, Reindel R, et al. The transcriptional profile of coronary arteritis in Kawasaki disease. BMC Genomics. 2015;16(1):1076.
Hamada H, Suzuki H, Onouchi Y, Terai M, Fuse S, Okajima Y, et al. Efficacy of primary treatment with immunoglobulin plus ciclosporin for prevention of coronary artery abnormalities in patients with Kawasaki disease predicted to be at increased risk of non-response to intravenous immunoglobulin (KAICA): a randomised controlled, open-label, blinded-endpoints, phase 3 trial. Lancet. 2019;393(10176):1128–37.
Burns JC. Cyclosporine and coronary outcomes in Kawasaki disease. J Pediatr. 2019;210:239–42.
Niedra E, Chahal N, Manlhiot C, Yeung RS, McCrindle BW. Atorvastatin safety in Kawasaki disease patients with coronary artery aneurysms. Pediatr Cardiol. 2014;35(1):89–92.
Peng S, Xu LW, Che XY, Xiao QQ, Pu J, Shao Q, et al. Atorvastatin inhibits inflammatory response, attenuates lipid deposition, and improves the stability of vulnerable atherosclerotic plaques by modulating autophagy. Front Pharmacol. 2018;9:438.
Tremoulet AH, Jain S, Jone P-N, Best BM, Duxbury EH, Franco A, et al. Phase I/IIa trial of atorvastatin in patients with acute Kawasaki disease with coronary artery aneurysm. The Journal of Pediatrics. 2019;215:107–17.e12.
Tremoulet AH, Jain S, Jone PN, Best BM, Duxbury EH, Franco A et al. Phase I/IIa trial of atorvastatin in patients with acute Kawasaki disease with coronary artery aneurysm. J Pediatr. 2019;215:107–17.e12.
Tang T-T, Song Y, Ding Y-J, Liao Y-H, Yu X, Du R, et al. Atorvastatin upregulates regulatory T cells and reduces clinical disease activity in patients with rheumatoid arthritis. J Lipid Res. 2011;52(5):1023–32.
Siber GR, Werner BG, Halsey NA, Reid R, Almeido-Hill J, Garrett SC, et al. Interference of immune globulin with measles and rubella immunization. J Pediatr. 1993;122(2):204–11.
Sonobe T. Intravenous gamma-globulin therapy and vaccination. Shoni-naika; in Japanese. 1994;26:1929–33.
Tacke CE, Smits GP, van der Klis FRM, Kuipers IM, Zaaijer HL, Kuijpers TW. Reduced serologic response to mumps, measles, and rubella vaccination in patients treated with intravenous immunoglobulin for Kawasaki disease. J Allergy Clin Immunol. 2013;131(6):1701–3.
Esposito S, Bianchini S, Dellepiane RM, Principi N. Vaccines and Kawasaki disease. Expert Rev Vaccines. 2016;15(3):417–24.
Funding
The work of M.A. is supported by the NIH Grant R01 AI072726 and M.N.R. is supported by the NIH grant R01 HL139766.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
No potential conflicts of interest relevant to this article were reported.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pediatric Rheumatology
Rights and permissions
About this article

Cite this article
Soni, P.R., Noval Rivas, M. & Arditi, M. A Comprehensive Update on Kawasaki Disease Vasculitis and Myocarditis. Curr Rheumatol Rep 22, 6 (2020). https://doi.org/10.1007/s11926-020-0882-1
Published:
DOI: https://doi.org/10.1007/s11926-020-0882-1