
Abstract
Purpose of Review
The purpose of the study is to briefly review the molecular mechanisms that leads to structural damage in ankylosing spondylitis (AS), defined as new bone formation resulting in complete or incomplete ankylosis of the spine, and the impact of treatment with biologics to retard this process.
Recent Findings
The understanding of molecular mechanisms leading to new bone formation in AS has significantly improved but is still incomplete. Availability of biologics has greatly enhanced the treatment of patients with AS, but its impact on slowing the structural damage is still a matter of debate, although a few observational studies have shown that long term use of TNF-α blockers may slow radiographic progression. The availability of newer biologics targeting IL-17/1L23 has shown some promising results in slowing radiographic progression in AS.
Summary
Although the availability of TNF-inhibitors has greatly enhanced the treatment options for patients with AS, their impact on slowing the structural damage is still not clearly established. However, preliminary results using newer biologics targeting IL-17/1L23 axis are more encouraging but longer follow-up is needed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ankylosing spondylitis (AS) is a chronic disease that is characterized by inflammation of the axial spine [1••, 2]. The consequences of bone inflammation in AS are increased bone resorption juxtaposed with osteoproliferation resulting in ossifying enthesitis and ankylosis of the sacroiliac joints and intervertebral discs [3]. The major goals of management of patients with AS are to treat inflammation and to stop the structural damage, defined as new bone formation resulting in complete or incomplete ankylosis of the spine. Our understanding of the molecular mechanisms leading to new bone formation in AS has significantly improved but still not complete. The availability of biologics has greatly enhanced the treatment options for patients with AS. However, whether they slow the structural damage or not is still a matter of debate. Few observational studies have shown that long-term use of tumor necrosis factor-alpha (TNF-α) blockers may slow radiographic progression. The availability of newer biologics targeting IL-17/1L23 has shown some promising results in terms of slowing radiographic progression in AS. In this review, we first briefly discuss the molecular mechanism leading to new bone formation in AS, and then review the therapies available to halt this process.
New Bone Formation in AS
The major hallmark of AS is syndesmophyte formation that leads to fusion of the spine. This is coupled with osteoporosis of the spine, thereby increasing the risk of vertebral fractures. It is still puzzling as to what leads to simultaneous bone loss with new bone formation. Although the distinctive characteristic of AS is sacroiliitis but enthesitis both in the axial and appendicular skeleton is the primary feature of the disease. Pathologically, AS can be divided into three-stages, acute inflammatory reaction at the fibrocartilagenous enthesis, leading to erosions which are followed by new bone formation or enthesopathy at the site [4]. There is radiographic and pathologic evidence supporting this view. The two molecular signaling pathways that have been implicated in new bone formation in AS are the bone morphogenetic proteins (BMPs) mediated pathway and Wnt signaling pathways, based on animal studies.
BMPs are a group of cytokines and growth factors that belong to the transforming growth factor β superfamily and are involved in bone formation. They bind to BMP receptors, activating both the canonical Smad-dependent signaling pathway and the non-canonical p38 mitogen-activated protein kinase pathway to regulate mesenchymal stem cell differentiation during skeletal development, bone formation, and bone homeostasis [5]. Noggin is an extracellular BMP antagonist which binds to BMPs 2, 4, 6, and 7 and prevents them from binding to their receptors [6]. Activation of BMP signaling has been studied in a mouse model of degenerative arthritis and specific BMPs (BMP2 and BMP6 and BMP7) were found to be involved in embryonic endochondral bone formation [7]. Activation of BMP signals was also confirmed in entheseal biopsies obtained from Achilles tendons of spondyloarthritis (SpA) patients. Noggin gene transfer was shown to ameliorate disease progression. However, this animal model lacked axial joint involvement.
BMP 6 polymorphisms have also been identified as possible risk factors for the development of syndesmophyte and ankylosis in AS [8]. A study of Korean patients with AS showed that two single nucleotide polymorphisms in BMP6 were significantly associated with radiologic severity in these patients [8]. Studies of the concentrations of BMPs in the sera of patients with AS have yielded conflicting results. Increased serum level of BMPs in patients with AS has been found in some studies [9], whereas other studies have not found such an increase [10].
Another possible mechanism for new bone formation in AS is abnormal osteogenic differentiation of bone marrow (BM) mesenchymal stem cells (MSC) that differentiate into bone tissue in vivo [11]. Alterations in the osteogenic differentiation of BM-MSCs contributing to rheumatic autoimmune diseases have been reported in some studies [12, 13]. The hypothesis that abnormal osteogenic differentiation of AS-MSCs could be the mechanism of pathologic osteogenesis in AS was tested in a recent study [14••]. The study revealed a mechanism of pathologic osteogenesis in AS by demonstrating an imbalance between BMP-2 and Noggin secretion in AS patients that possibly leads to abnormal osteogenic differentiation of AS MSCs.
MicroRNAs may play a role in new bone formation in SpA. MicroRNAs (miRNAs) are small ∼22 nucleotide long non-coding RNAs that post transcriptionally regulate gene expression by targeting specific messenger RNAs (mRNAs) for degradation or translational repression. miRNAs play a role in various immune pathways and regulate the function of both the innate and the adaptive immune systems. Differentially expressed miRNAs have also been identified in SpA and miR-34a which was overexpressed in patients with axial SpA compared to healthy controls and was predicted to target BMP-3 [15••]. miR-34a has also been found to be a novel and critical suppressor of osteoclastogenesis.
In addition to BMPs triggering new bone formation in AS, Wnt signaling pathways have emerged as critical pathways in osteoblastic bone formation [16] and play a role in bone formation in AS. Natural inhibitors of the Wnt signaling pathway Dickkopf-related protein 1 (DKK1) and sclerostin have been linked to bone formation in AS [17]. Blockade of DKK1 by a neutralizing antibody was shown to promote ankylosis of the sacroiliac joints in human TNF transgenic mice [18]. Low levels of DKK-1 have been found in patients with AS compared to healthy controls [19]. High levels of DKK-1 were shown to protect patients with AS from syndesmophyte formation and a high correlation was found between DKK-1 and sclerostin levels in patients with radiographic progression [20]. Moreover, serum levels of sclerostin have been found to be lower in patients with AS than in healthy individuals and these low levels in patients with AS were significantly associated with the formation of new syndesmophytes [21].
It is well established that mechanotransduction is critical in maintaining bone strength and quality under physiological conditions. Mechanical loading (ML) of the bone mediates activation of signaling pathways that result in cell differentiation and bone formation [22]. This has been conceptualized as a possible mechanism of new bone formation in AS but the evidence is limited. A recent study suggested that new bone formation in SpA may be influenced by ML. The study revealed that in a collagen antibody-induced arthritis mouse model (CAIA), osteophytes were significantly smaller when weight bearing was prevented, supporting a role for mechanical strain in their development [23].
Role of Inflammation in Osteogenesis in AS
Inflammation has been shown to trigger bone loss. Pro-inflammatory cytokines involved in inflammation can affect osteoclast and osteoblast activity, leading to systemic bone loss [24••, 25]. Pathways activated by cytokines like TNF-α and transcription factor-like receptor activator of nuclear factor kappa ligand (NF-κB) play an essential role in inflammation and also in upregulation of osteoclastogenesis, resulting in bone loss in AS [26, 27]. However, the role of inflammation on new bone formation in AS is not well understood [28].
It has been hypothesized that new bone formation in patients with AS may be independent of inflammation and continues even after the inflammation has been resolved [29]. In a mouse model of SpA, blocking of TNF-α with etanercept did not inhibit the formation of new cartilage and bone at the enthesis [30]. This concept was supported by clinical studies that showed use of infliximab, adalimumab, or etanercept for up to 2 years did not slow radiographic progression in patients with AS [31,32,33] suggesting that structural damage in AS may be independent of inflammation and the effects of TNF. Various important biases have been identified in these studies which include short duration of the study, use of a historical cohort for comparison, and the relatively low sensitivity of the outcome parameters of radiographic progression (as assessed by the modified Stoke Ankylosing Spondylitis Spine Score [mSASSS]).
The other plausible explanation for this observation is that TNF-α upregulates DKK1 and sclerostin, thereby inhibiting the Wnt-Frizzled pathway and also new bone formation. If valid, the use of TNF-α inhibitors in AS will upregulate the Wnt pathway by downregulating its inhibitors and resulting in new bone formation. However, this has not been observed clinically. A retrospective study that compared the rate of radiographic progression in patients with AS treated with infliximab (INF) over an 8-year period to a historical cohort of patients with AS who had never been treated with TNF-α blockers found that new bone formation was seen in both groups [34]. The rate of new bone formation over time was not increased in the continuous anti-TNF therapy group compared to the historical cohort. The number of syndesmophytes, although similar at baseline, differed significantly at 8 years: 1.0 ± 0.6 new syndesmophytes/patient in infliximab versus 2.7 ± 0.8 in the historical cohort (p = 0.007) [32], suggesting that prolonged use of infliximab slows radiographic progression.
Paradoxical Effect of TNF-α on Bone Formation in AS
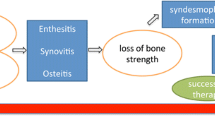
Long-term use of TNF-α inhibitors has shown decreased progression of new bone formation in AS, suggesting a possible paradoxical effect of TNF-α on bone formation in AS as shown in Fig. 1 [34, 35••]. Recently, some murine and human studies have shown that TNF-α may have a mixed effect on bone homeostasis and may actually trigger osteoproliferation and new bone formation, depending upon the local milieu and concentration of TNF. TNF-α has been shown to increase BMP-2 expression in human MSCs through the NF-κB signaling pathway in early osteogenic differentiation [36••]. NF-κB stimulates critical regulators of osteogenesis like BMP2, RUNX2, and Osterix resulting in enhanced mineralization of the extracellular matrix [37]. Low concentrations of TNF-α have been shown to increase osteogenic differentiation by upregulation of Runx2, osteocalcin, and alkaline phosphatase levels in murine MSC studies [38].

Schematic representation linking hypothesis of disease predisposition, immune activation and cytokine release leading to bone loss and new bone formation in ankylosing spondylitis. BM2 bone morphogenetic protein 2, RUNX2 runt-related transcription factor-2, hMSCs human mesenchymal stem cells
Another possible explanation of the osteogenic effect of TNF-α in AS is that at the site of enthesitis where syndesmophytes usually develop, it is plausible that osteocytes may not be in direct contact with tendon-derived osteoblasts and TNF-α may thereby exhibit an osteogenic effect there [36••]. Also, the DKK1 levels were seen to be increased in AS patients with anti-TNF-α treatment [39]. This effect is in complete contrast to what has been seen in rheumatoid arthritis where the classic effect of anti-TNF-α therapy that reduces DDK1 is observed [39].
Inflammation May Trigger New Bone Formation in AS
Evidence has been accumulating in recent years suggesting that inflammation may trigger new bone formation in AS. In a B27/hβ(2) m-transgenic SpA mouse model, vertebral samples from rats with minimal or absent inflammation revealed no osteoproliferation. However, osteoblastic activity was shown to be present at the edge of the vertebrae in sections with moderate inflammation that persisted during severe inflammation and end-stage destruction [40]. The evidence that inflammation triggers new bone formation in AS is mostly observational and is based on clinical and MRI studies. Clinical studies have associated new bone formation to elevated markers of inflammation (for the erythrocyte sedimentation rate, odds ratio [OR] = 4.04, p = 0.001; for C-reactive protein level time-averaged over 2 years, OR = 3.81, p = 0.001) [41]. MRI studies have linked new bone formation to inflammatory lesions of the spine [42••, 43]. The studies suggest that presence of “fatty lesions”, detected as hyperintense signals due to low water content and similarity to the fat tissue on the T1-weighted MRI sequence are considered as early chronic changes [44]. The combination of acute inflammation on STIR sequence (fat-suppressed sequence) and fatty lesions have been found to be most predictive of syndesmophyte formation in AS [42••]. Fat metaplasia is considered an intermediate step towards new bone formation in AS. However, a recent in situ analysis of subchondral granulation tissue in the facet joints of AS patients revealed direct invasion of the granulation tissue into the subchondral bone, suggesting that granulation tissue may have a pivotal role in progressive ankylosis in AS [45••].
Based on this information, it has been proposed that if early inflammatory lesions resolve without undergoing chronic changes, the sequelae of new bone formation following inflammation might be halted. Hence, it has been hypothesized that there may be a window of opportunity in the treatment of AS and that early treatment with anti-TNF therapy may halt inflammation and new syndesmophyte formation. This was tested prospectively in a study of 76 AS patients recruited to a placebo-controlled trial of adalimumab therapy for a period of 104 weeks [46]. The study revealed that the majority of new syndesmophytes (26/48 (54.2%)) occurred at those vertebral corners that had either a fat lesion and/or a chronic inflammatory lesion on baseline MRI. The odds of developing syndesmophytes were much higher in patients with chronic inflammatory lesions (OR = 3.88; 95% CI [1.20–12.57], p = 0.024) or a fat lesion (OR = 4.83; 95% CI [2.38 to –9.80], p < 0.0001). This information has prompted studying whether radiographic progression in AS can be stopped by the use of biologic therapy.
Do Biologics Slow Radiographic Progression in AS?
Use of biologics has revolutionized the treatment of AS. TNF-α inhibitor therapy has been recommended for the treatment of patients with AS who fail to respond to traditional non-steroidal anti-inflammatory agents (NSAIDs) [47••, 48]. There is overwhelming evidence about the efficacy and safety of long-term use of TNF-α inhibitors in AS [49, 50]. However, the efficacy of anti-TNF-α therapy on radiographic progression is uncertain. Even though short-term studies with anti-TNF-α therapy in AS have failed to slow the radiographic progression, a few long-term studies have shown some promising results.
As mentioned earlier, a small retrospective study revealed that prolonged use of infliximab over a period of 8 years slowed new syndesmophyte formation in patients with AS [34]. Moreover, a recent prospective longitudinal observational cohort study in daily clinical practice showed overall slow and linear spinal radiographic progression in AS patients with long-term TNF-α blocking therapy [51]. The study enrolled 201 consecutive patients from the Groningen Leeuwarden AS (GLAS) cohort that had initiated treatment with TNF-alpha inhibitors during 2004–2012 and had received baseline and biannual radiographs over an 8-year follow-up. Spinal radiographic progression as measured by mSASSS over 8 years of follow-up declined steadily to 1.4 points in years 2–4 from 2.4 in the first 2 years, 1.0 in years 4–6, and 0.8 in years 6–8. The authors concluded that long-term inhibition of inflammation with TNF-alpha inhibitors may diminish new bone formation over time in patients with long-standing AS [51, 52••].
Similar results were also observed in another longitudinal cohort study in which patients were followed at five different centers in North America [35••]. Patients received TNF-α inhibitors as the standard of care if disease activity was not controlled by NSAIDs. TNF-α inhibitor use was associated with a 50% decreased odds of progression of radiographic damage. Radiographic disease progression was slower in patients in whom TNF-blocker therapy was started earlier in the course of disease than in patients in whom start of treatment was delayed. These data suggest that early and long-term treatment of bone inflammation with TNF blockers can prevent radiographic disease progression.
The fundamental assumption from these observational studies is that there has been no unmeasured confounding as sensitivity analysis was done to interpret the results. However, it is known that radiographic progression in AS is overall slow and highly variable between patients, so different patient numbers at different points during the follow-up may have affected the outcome. Also, radiographic progression as measured by mSASSS may not have be a very precise measurement of change.
Results of a long-term study in patients with axial SpA, including patients with AS and non-radiographic SpA, treated with certolizumab pegol, revealed minimal radiographic progression in the first 4 years of treatment [53]. Of 315 certoluzimuab treated patients, 196 had available spinal X-rays and were included in the analysis. In patients with AS, mean mSASSS change between baseline and week 204 was 0.98 (95% CI: 0.34–1.63). This is the first report of a 4-year imaging data from a clinical trial; however, all patients analyzed received certolizumab so no comparison arm is available. Whether TNF inhibitors limit the development of new radiographic damage continues to remain a matter of debate and data from long-term randomized placebo controlled trials are needed to answer the question and that may not be feasible.
Role of IL-17 Inhibitors in Slowing Radiographic Progression in AS
The IL-23/IL-17/IL-22 axis has emerged as a critical pathway in the pathogenesis of AS and new biologic therapies are being developed to target this pathway [54,55,56,57]. IL-23 signaling promotes CD 4+ Th17 cell differentiation, resulting in increased IL-17A production [58]. The IL-23/IL-17 axis, besides invoking synovial inflammation and joint erosion, also plays a critical role in new bone formation. Special entheseal CD4 and CD8 negative resident T cells [γδ CD3 +] have been detected in a mouse model that respond to IL-23 in vitro and elaborate inflammatory mediators including IL-6, IL-17, IL-22, and chemokine (C-X-C motif) ligand 1 (CXCL1) [53]. Overproduction of IL-22 has been associated with transcriptomic signatures of osteoproliferation and new bone formation.
The impact of anti-interleukin 17A (IL-17A) antibody on new bone formation was assessed in a validated animal model of SpA. Rats (n = 6) were treated weekly with an anti-mouse/rat IL-17A antibody. Micro CT and histology data indicated that IL-17A blockade reduced structural damage, including pathological new bone formation in the rats [59]. The effect of IL-17A antibody on radiographic progression was studied in a recent randomized controlled trial in AS patients. The results from the international phase III MEASURE 1 study showed promising low mean progression rates in spinal radiographic change with the anti-interleukin 17A antibody, secukinumab in patients with AS [60••]. This was a 2-year study where 246 adult AS patients were randomized to receive either placebo or secukinumab. Among patients with evaluable X-rays who were originally randomized to receive secukinumab (n = 168), the mean change in the mSASSS from baseline to week 104 was 0.30 ± 2.53. About 62% (n = 104) of these patients had baseline syndesmophytes ≤0. Patients with no syndesmophytes at baseline and who were randomized to secukinumab, 61/64 (95.3%), remained free from syndesmophytes at week 104. The low overall rate of progression seen with secukinumab definitely needs further exploration in long-term controlled studies.
Effect of NSAIDs on New Bone Formation
NSAIDs have been traditionally used for ameliorating pain and stiffness in AS but slowing of radiographic progression with NSAIDs was also revealed early on in 1976 [61] and has been confirmed in randomized clinical trials subsequently [62, 63]. The inhibitory effect of NSAIDs on bone formation has been linked to their capacity to inhibit cyclooxygenase (COX) activity and consequent prostaglandin synthesis. Genome-wide association studies have found a strong association between the gene PTGER4 and AS [64]. This gene encodes for prostaglandin E receptor 4, one of four receptors for prostaglandin E2 (PGE2), which modulate osteoblastic and osteoclastic function under physiological or pathological conditions and is inhibited by NSAIDs. A recent clinical trial over a 2-year period, however, could not confirm the disease modifying effect of NSAIDs, adding to the enigma of new bone formation in AS [65••] (Fig. 1).
Conclusion
Our understanding of the mechanisms underlying new bone formation in AS has improved but is certainly not complete. There is evidence that bone formation is triggered by the inflammatory response, but some degree of independence between the inflammation and the new bone formation is also evident in the latter stages of the disease. Early and prolonged use of TNF-α inhibitors may slow radiographic progression either by themselves or in combination with NSAIDs. The newer biologics targeting IL-17/1L23 have shown some promising results in terms of slowing radiographic progression in AS. Long-term controlled studies are needed to verify these findings.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
•• Taurog JD, Chhabra A, Colbert RA. Ankylosing spondylitis and axial spondyloarthritis. N Engl J Med. 2016;374:2563–74. This review article highlights recent advances in our understanding of relationship between spondyloarthritis and ankylosing spondylitis and treatments available.
Khan MA. Ankylosing spondylitis and related spondyloarthropathies: the dramatic advances in the past decade. Rheumatology (Oxford). 2011;50:637–9.
Appel H, Maier R, Loddenkemper C, et al. Immunohistochemical analysis of osteoblasts in zygapophyseal joints of patients with ankylosing spondylitis reveal repair mechanisms similar to osteoarthritis. J Rheumatol. 2010;37:823–8.
Lories RJ, Schett G. Pathophysiology of new bone formation and ankylosis in spondyloarthritis. Rheum Dis Clin N Am. 2012;38:555–67.
Biver E, Hardouin P, Caverzasio J. The “bone morphogenic proteins” pathways in bone and joint diseases: translational perspectives from physiopathology to therapeutic targets. Cytokine Growth Factor Rev. 2013;24:69–81.
Krause C, Guzman A, Knaus P. Noggin. Int J Biochem Cell Biol. 2011;43:478–81.
Lories RJU, Derese I, Luyten FP. Modulation of bone morphogenetic protein signaling inhibits the onset and progression of ankylosing enthesitis. J Clin Invest. 2005;115:1571–9.
Joo YB, Bang SY, Kim TH, Shim SC, Lee S, et al. Bone morphogenetic protein 6 polymorphisms are associated with radiographic progression in ankylosing spondylitis. PLoS One. 2014;9:e104966.
Chen HA, Chen CH, Lin YJ, Chen PC, Chen WS, Lu CL, et al. Association of bone morphogenetic proteins with spinal fusion in ankylosing spondylitis. J Rheumatol. 2010;37:2126–32.
Wendling D, Cedoz JP, Racadot E, Dumoulin G. Serum IL-17, BMP-7, and bone turnover markers in patients with ankylosing spondylitis. Joint Bone Spine. 2007;74:304–5.
El TM, Reis RL. Progenitor and stem cells for bone and cartilage regeneration. J Tissue Eng Regen Med. 2009;3:327–37.
Mohanty ST, Kottam L, Gambardella A, Nicklin MJ, Coulton L, Hughes D, et al. Alterations in the self-renewal and differentiation ability of bone marrow mesenchymal stem cells in a mouse model of rheumatoid arthritis. Arthritis Res Ther. 2010;12:R149.
Sun LY, Zhang HY, Feng XB, Hou YY, Lu LW, Fan LM. Abnormality of bone marrow-derived mesenchymal stem cells in patients with systemic lupus erythematosus. Lupus. 2007;16:121–8.
•• Xie Z, Wang P, Li Y, Deng W, Zhang X, et al. Imbalance between bone morphogenetic protein 2 and noggin induces abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. Arthritis Rheum. 2016;68:430–40. The study revealed a mechanism of pathologic osteogenesis in AS by demonstrating an imbalance between BMP-2 and Noggin secretion in AS patients that possibly leads to abnormal osteogenic differentiation of AS MSCs.
•• Magrey MN, Haqqi T, Haseeb A. Identification of plasma microRNA expression profile in radiographic axial spondyloarthritis—a pilot study. Clin Rheumatol. 2016;35:1323–7. Brief report about microRNA expression in AS and microRNA 34a is differentially expressed in SpA and has been shown to inhibit bone loss.
Goldring SR, Goldring MB. Eating bone or adding it: the Wnt pathway decides. Nat Med. 2007;13:133–4.
Xie W, Zhou L, Li S, Hui T, Chen D. Wnt/β-catenin signaling plays a key role in the development of spondyloarthritis. Ann N Y Acad Sci. 2016;1364:25–31.
Uderhardt S, Diarra D, Katzenbeisser J, David JP, Zwerina J, et al. Blockade of Dickkopf (DKK)-1 induces fusion of sacroiliac joints. Ann Rheum Dis. 2010;3:592–7.
Kwon SR, Lim MJ, Suh CH, Park SG, Hong YS, Yoon BY, et al. Dickkopf-1 level is lower in patients with ankylosing spondylitis than in healthy people and is not influenced by anti-tumor necrosis factor therapy. Rheumatol Int. 2012;32:2523–7.
Heiland GR, Appel H, Poddubnyy D, Zwerina J, Hueber A, et al. High level of functional dickkopf-1 predicts protection from syndesmophyte formation in patients with ankylosing spondylitis. Ann Rheum Dis. 2012;71:572–4.
Appel H, Ruiz-Heiland G, Listing J, Zwerina J, Herrmann M, et al. Altered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondylitis. Arthritis Rheum. 2009;60:3257–62.
González-Chávez SA, Quiñonez-Flores CM, Pacheco-Tena C. Molecular mechanisms of bone formation in spondyloarthritis. Joint Bone Spine. 2016;83:394–400.
Jacques P, Lambrecht S, Verheugen E, Pauwels E, Kollias G, Armaka M, et al. Proof of concept: enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann Rheum Dis. 2014;73:437–45.
•• Shaw AT, Gravallese EM. Mediators of inflammation and bone remodeling in rheumatic disease. Semin Cell Dev Biol. 2016;49:2–10. This review article discusses the effects of cytokines on bone in two settings, rheumatoid arthritis and spondyloarthritis.
Walsh NC, Reinwald S, Manning CA, et al. Osteoblast function is compromised at sites of focal bone erosion in inflammatory arthritis. J Bone Miner Res. 2009;24:1572–85.
Braun J, Bollow M, Neure L, Seipelt E, Seyrekbasan F, Herbst H, et al. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis Rheum. 1995;38:499–505.
Im CH, Kang EH, Ki JY, et al. Receptor activator of nuclear factor kappa B ligand-mediated osteoclastogenesis is elevated in ankylosing spondylitis. Clin Exp Rheumatol. 2009;27:620–5.
Sieper J, Poddubnyy D. Inflammation, new bone formation and treatment options in axial spondyloarthritis. Ann Rheum Dis. 2014;73:1439–41.
Pedersen SJ, Sorensen IJ, Lambert RG, Hermann KG, Garnero P, Johansen JS, et al. Radiographic progression is associated with resolution of systemic inflammation in patients with axial spondylarthritis treated with tumor necrosis factor a inhibitors: a study of radiographic progression, inflammation on magnetic resonance imaging, and circulating biomarkers of inflammation, angiogenesis, and cartilage and bone turnover. Arthritis Rheum. 2011;63:3789–800.
Lories RJ, Derese I, de Bari C, Luyten FP. Evidence for uncoupling of inflammation and joint remodeling in a mouse model of spondylarthritis. Arthritis Rheum. 2007;56:489–97.
van der Heijde D, Salonen D, Weissman BN, Landewé R, Maksymowych WP, Kupper H, et al. Assessment of radiographic progression in the spines of patients with ankylosing spondylitis treated with adalimumab for up to 2 years. Arthritis Res Ther. 2009;11:R127.
van der Heijde D et al. Radiographic findings following two years of infliximab therapy in patients with ankylosing spondylitis. Arthritis Rheum. 2008;58:3063–70.
van der Heijde D, Landewé R, Einstein S, Ory P, Vosse D, Ni L, et al. Radiographic progression of ankylosing spondylitis after up to two years of treatment with etanercept. Arthritis Rheum. 2008;58:1324–31.
Baraliakos X, Haibel H, Listing J, Sieper J, Braun J. Continuous long-term anti-TNF therapy does not lead to an increase in the rate of new bone formation over 8 years in patients with ankylosing spondylitis. Ann Rheum Dis. 2014;73:710–5.
•• Haroon N, Inman RD, Learch TJ, et al. The impact of tumor necrosis factor alpha inhibitors on radiographic progression in ankylosing spondylitis. Arthritis Rheum. 2013;65:2645–54. This observational study shows that long term use of TNF inhibitors decreases the odds of radiographic progression in AS.
•• Osta B, Benedetti G, Miossec P. Classical and paradoxical effects of TNF-α on bone homeostasis. Front Immunol. 2014;5:48. A thorough review about the effect on TNF-α on bone homeostasis.
Hess K, Ushmorov A, Fiedler J, Brenner RE, Wirth T. TNF alpha promotes osteogenic differentiation of human mesenchymal stem cells by triggering the NF-kappa B signaling pathway. Bone. 2009;45:367–76.
Huang H, Zhao N, Xu X, Xu Y, Li S, Zhang J. Dose-specific effects of tumor necrosis factor alpha on osteogenic differentiation of mesenchymal stem cells. Cell Prolif. 2011;44:420–7.
Daoussis D, Liossis SN, Solomou EE, Tsanaktsi A, Bounia K, Karampetsou M, et al. Evidence that Dkk-1 is dysfunctional in ankylosing spondylitis. Arthritis Rheum. 2010;62:150–8.
van Duivenvoorde LM, Dorris ML, Satumtira N, van Tok MN, Redlich K, Tak PP, et al. Relationship between inflammation, bone destruction, and osteoproliferation in the HLA-B27/human β2-microglobulin-transgenic rat model of spondylarthritis. Arthritis Rheum. 2012;64:3210–9.
Poddubnyy D, Haibel H, Listing J, Märker-Hermann E, Zeidler H, Braun J, et al. Baseline radiographic damage, elevated acute-phase reactant levels, and cigarette smoking status predict spinal radiographic progression in early axial spondylarthritis. Arthritis Rheum. 2012;64:1388–98.
•• Baraliakos X, Heldmann F, Callhoff J, Listing J, Appelboom T, Brandt J, et al. Which spinal lesions are associated with new bone formation in patients with ankylosing spondylitis treated with anti-TNF agents? A long-term observational study using MRI and conventional radiography. Ann Rheum Dis. 2014;73:1819–25. The study revealed that combination of acute inflammation on STIR sequence (fat-suppressed sequence) and fatty lesions are most predictive of syndesmophyte formation in AS.
Maksymowych WP, Chiowchanwisawakit P, Clare T, et al. Inflammatory lesions of the spine on magnetic resonance imaging predict the development of new syndesmophytes in ankylosing spondylitis: evidence of a relationship between inflammation and new bone formation. Arthritis Rheum. 2009;60:93–102.
Rudwaleit M, Jurik AG, Hermann KG, Landewe R, van der Heijde D, Baraliakos X, et al. Defining active sacroiliitis on magnetic resonance imaging (MRI) for classification of axial spondyloarthritis:a consensual approach by the ASAS/OMERACT MRI group. Ann Rheum Dis. 2009;68:1520–7.
•• Bleil J, Maier R, Hempfing A, Sieper J, Appel H, Syrbe U. Granulation tissue eroding the subchondral bone also promotes new bone formation in ankylosing spondylitis. Arthritis Rheum. 2016;68:2456–65. Granulation tissue and not fatty metaplasia may be promoting new bone formation in AS.
Maksymowych WP, Morency N, Conner-Spady B, Lambert RG. Suppression of inflammation and effects on new bone formation in ankylosing spondylitis: evidence for a window of opportunity in disease modification. Ann Rheum Dis. 2013;72:23–8.
•• Ward MM, Deodhar A, Akl EA, Lui A, Ermann J, Gensler LS, et al. American college of rheumatology/spondylitis association of america/spondyloarthritis research and treatment network 2015 recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Care Res (Hoboken). 2016;68:151–66. ACR/SPARTAN recommendation for treatment of AS.
Rohekar S, Chan J, Tse SM, Haroon N, Chandran V, Bessette L, et al. 2014 update of the Canadian rheumatology association/spondyloarthritis research consortium of Canada treatment recommendations for the management of spondyloarthritis. Part II: specific management recommendations. J Rheumatol. 2015;42:665–81.
Baraliakos X, van den Berg R, Braun J, van der Heijde D. Update of the literature review on treatment with biologics as a basis for the first update of the ASAS/EULAR management recommendations of ankylosing spondylitis. Rheumatology (Oxford). 2012;51:1378–87.
Sieper J, Poddubnyy D. New evidence on the management of spondyloarthritis. Nat Rev Rheumatol. 2016;12:282–95.
Maas F, Spoorenberg A, Brouwer E, Bos R, Efde M, Chaudhry RN, et al. Spinal radiographic progression in patients with ankylosing spondylitis treated with TNF-α blocking therapy: a prospective longitudinal observational cohort study. PLoS One. 2015;10(4):e0122693.
•• Maas F, Arends S, Brouwer E, Essers I, van der Veer E, Efde M, et al. Reduction in spinal radiographic progression in ankylosing spondylitis patients receiving prolonged treatment with TNF-α inhibitors. Arthritis Care Res (Hoboken). 2016. doi:10.1002/acr.23097. A longitudinal observational cohort study in daily clinical practice showed an overall slow and linear spinal radiographic progression in AS patients with long-term TNF-α blocking therapy.
van der Heijde D, Baraliakos X, Hermann KG, Landewé R, Machado P, Maksymowych W, Davies O, de Peyrecave N, Hoepken B, Bauer L, Nurminen T, Braun J. Four year imaging outcomes in patients with axial spondyloarthritis treated with certolizumab pegol, including patients with ankylosing spondylitis and non-radiographic axial spondyloarthritis [abstract]. Arthritis Rheumatol. 2016; 68 (suppl 10).
Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. 2012;18:1069–76.
Smith JA, Colbert RA. The IL-23/IL-17 axis in spondyloarthritis pathogenesis: Th17 and beyond. Arthritis Rheum. 2014;66:231–41.
Appel H, Maier R, Wu P, Scheer R, Hempfing A, Kayser R, et al. Analysis of IL- 17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res Ther. 2011;13:R95.
Shen H, Goodall JC, Hill Gaston JS. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. 2009;60:1647–56.
McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–24.
van Tok M, van Duivenvoorde L, et al. Anti-IL17A treatment blocks new bone formation in experimental spondyloarthritis in HLA-B27 transgenic rats. Arthritis Rheumatol. 2015; 67 (suppl 10) [abstract].
•• Braun J, Baraliakos X, Deodhar A, Baeten D, Sieper J, Emery P, Readie A, Martin R, Mpofu S, Richards HB. Effect of secukinumab on clinical and radiographic outcomes in ankylosing spondylitis: 2-year results from the randomised phase III MEASURE 1 study. Ann Rheum Dis. 2016 Dec 13. The paper describes the results from the international phase III MEASURE 1 study which showed low mean progression rates in spinal radiographic change with the anti-interleukin 17A antibody, secukinumab in patients with AS.
Boersma JW. Retardation of ossification of the lumbar vertebral column in ankylosing spondylitis by means of phenylbutazone. Scand J Rheumatol. 1976;5:60–4.
Wanders A, Heijde D, Landewe R, et al. Nonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis: a randomized clinical trial. Arthritis Rheum. 2005;52:1756–65.
Poddubnyy D, Rudwaleit M, Haibel H, et al. Effect of nonsteroidal anti-inflammatory drugs on radiographic spinal progression in patients with axial spondyloarthritis: results from the German Spondyloarthritis Inception Cohort. Ann Rheum Dis. 2012;71:1616–22.
Lories RJ, Haroon N. Bone formation in axial spondyloarthritis. Best Pract Res Clin Rheumatol. 2014;28:765–77.
•• Sieper J, Listing J, Poddubnyy D, et al. Effect of continuous versus on-demand treatment of ankylosing spondylitis with diclofenac over 2 years on radiographic progression of the spine: results from a randomized multicenter trial (ENRADAS). Ann Rheum Dis. 2016;75:1438–43. The study could not confirm the disease modifying effect of NSAIDs in AS.
Acknowledgements
We would like to thank Dr. Irving Kushner, Dr. Stanley Ballou, and Dr. Maria Antonelli for reviewing the paper and making necessary edits.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Magrey reports personal fees from UCB PHARMA and personal fees from Jansenn, outside the submitted work.
Dr. Khan declares no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors
Additional information
This article is part of the Topical Collection on Spondyloarthritis
Rights and permissions
About this article

Cite this article
Magrey, M.N., Khan, M.A. The Paradox of Bone Formation and Bone Loss in Ankylosing Spondylitis: Evolving New Concepts of Bone Formation and Future Trends in Management. Curr Rheumatol Rep 19, 17 (2017). https://doi.org/10.1007/s11926-017-0644-x
Published:
DOI: https://doi.org/10.1007/s11926-017-0644-x