
Abstract
Osteoarthritis (OA), the most common type of arthritis worldwide, is a degenerative disease of diarthrodial joints resulting in pain, reduced quality of life, and socioeconomic burden. Gout, the most common form of inflammatory arthritis, is a consequence of persistently elevated levels of urate and the formation of proinflammatory monosodium urate crystals in joints. Clinicians have long noted a predilection for both diseases to occur in the same joints. In this review, we provide an overview into research elucidating possible biochemical, mechanical, and immunological relationships between gout and OA. We additionally consider the potential implications of these relationships for OA treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA), a progressive disease of diarthrodial joints, results in joint pain, functional limitation, morbidity, and socioeconomic burden [1]. Often but erroneously considered strictly a condition of “wear-and-tear,” OA is highly prevalent in individuals aged 50 and older [2]. Knee OA alone affects more than 250 million people worldwide [1]. Gout results from persistently elevated levels of serum urate (sUA) exceeding the saturation point for monosodium urate (MSU) crystallization, initiating subsequent joint inflammation [3•]. According to the 2007–2008 National Health and Nutrition Examination Survey (NHANES), 3.9 % of a representative sample of the US population reported that they had gout, indicating that as many as 12 million Americans may have the disease [4]. OA, the most common arthritis overall, and gout, the most common inflammatory arthritis, frequently co-occur in the individual patient [5].
Whether a pathogenic relationship links OA and gout remains unknown. Acute gouty attacks and tophi both occur more frequently in joints that are affected by OA [6], suggesting a connection, but not addressing causality. A relationship in which gout promotes the development and/or progression of OA would be particularly meaningful for OA research. While there are presently no agents approved for OA disease modification, several currently available, effective gout treatment agents could potentially be repurposed for OA trials, should a biological rationale be supported. Broadly speaking, a number of putative mechanisms exist to explain the apparent predilections of common target joints for both gout and OA. OA cartilage damage may promote MSU deposition, gouty involvement may predispose joints to OA, or both conditions may be the trigger for, or consequence of, shared inflammatory cascades. Here, we review the literature supporting the possible mechanisms that underlie the long-described clinical observations linking OA and gout.
Association Between OA and Gout
Clinical and Epidemiological Evidence
Primary OA is typically a disease of the middle-aged and elderly. Prevalence of OA in persons age 20 and older is estimated to be around 4.5 % for men and 7.3 % for women; the prevalence increases to 17 % for men and 29.6 % for women in individuals over age 60 [7]. Although OA affects both sexes, it is more common in women [8]. Risk factors for OA include being overweight or obese [9]. In addition, diabetes is considered a risk factor that may augment OA development and progression [10].
Like OA, gout is associated with advancing age, but unlike OA, it has a tendency to affect more men than women at all ages [3•]. However, the risk of developing gout increases sharply for women after menopause [3•]. Hyperuricemia is the most significant risk factor and a prerequisite for gout. It is important to note that not everyone with hyperuricemia develops gout, suggesting a more complex pathogenic link. Indeed, hyperuricemia is much more common than gout, with a reported prevalence of 21.4 % [11]. Similar to OA, both hyperuricemia and gout have been associated with obesity, metabolic syndrome, and type 2 diabetes [12].
A number of cohort and population studies have examined the association between gout and OA. Howard et al. assessed well-defined cohorts of older patients who had gout, hyperuricemia, or neither for clinical and radiographic knee OA, and found that the presence of gout was accompanied by a significantly higher prevalence of knee OA compared to controls (68 vs. 28 %) [13]. Moreover, patients in the gout group had higher mean Kellgren-Lawrence grades compared to controls, signifying greater radiographic OA severity. Patients with hyperuricemia demonstrated intermediate prevalence and severity of OA between the gout and control groups (52 % meeting criteria for knee OA), suggesting that the association between OA and gout may be mediated, at least in part, by elevated sUA levels as opposed to gout per se [13]. Kuo et al. conducted a large-scale case-control study of roughly 80,000 subjects and found that the presence of OA was associated with a higher risk for future gout, with an odds ratio of 1.27. Conversely, among individuals without OA at baseline, the presence of gout was significantly associated with elevated 1-, 2-, 5-, and 10-year risk of OA development, with an overall adjusted hazard ratio of 1.45 [14]. A cross-sectional study of more than 4000 participants showed that elevated sUA levels and the presence of hyperuricemia were significantly associated with knee osteophytes, a hallmark of OA, in female patients. This finding held true even after controlling for confounding factors, including the presence of diabetes and alcohol use [15].
Joints Affected by Gout and OA
Clinicians have long observed that joints with a predilection for gout, including both acute attacks and tophus formation, are among those most commonly affected by OA [6]. Using a self-report questionnaire, Roddy et al. performed a case-control survey assessing whether nodal OA (OA of the distal or proximal interphalangeal joints of the hands, accompanied by Heberden’s nodes) or OA of other sites is more prevalent in gout patients compared to healthy controls. They observed significant associations between gout and OA of the knee and/or hallux, but not with nodal OA [16]. However, the study was underpowered, leading the authors to suggest that their inability to identify a statistically significant association between gout and nodal OA may have resulted from type II error. Several other studies have demonstrated an association with regard to the first metatarsophalangeal (MTP), mid-foot, knee, and finger distal interphalangeal (DIP) joints [5]. In a hospital-based study of 262 patients with gout, Kawanoski-Minc et al. demonstrated an association between a history of acute gouty attacks and radiographic evidence of OA at the first MTP joints, tarsal joints, and knees [17]. Roddy et al. conducted a similar study among 359 patients in the community, using an initial postal questionnaire followed by a clinical assessment. The authors found a significant association between the acute gout and the presence of OA at the first MTP joint, mid-foot, knee, and DIP joints, even after adjusting for confounding variables [18]. The strength of the correlation between gout and OA was not changed by the chronicity of gout, leading the authors to suggest that the presence of OA may predispose the joints to acute gouty attacks rather than the converse [18]. However, other data, as discussed below, may support an inverted causal relationship.
OA as a Possible Risk Factor for Gout
Gout begins with the metabolic condition of hyperuricemia. In turn, hyperuricemia results in crystal deposition in joints, which triggers acute inflammatory attacks and promotes chronic tophus formation. While no evidence suggests that the local articular phenomena of OA affect systemic sUA levels, investigators have proposed that the local environment of the OA joint may create a salutary environment for crystal formation [5]. Several OA-related mechanisms have been proposed to explain this association, including biomechanical stress driving local urate production by chondrocytes, transient fluctuations in the concentration of UA in OA synovial fluid, and changes in synovial proteoglycans and cartilage integrity [5] (Fig. 1). Thus, the majority of the literature in this area focuses on local, intra-articular factors as opposed to systemic or genetic predispositions associated with OA.

Interactions between OA and gout in a diarthrodial joint. Illustrated is the possible role of OA cartilage fibrillation, providing a nidus for monosodium urate (MSU) crystal deposition, with the potential for further cartilage damage by MSU. Additionally, shedding of chondroitin sulfate from the cartilage into the synovial fluid may adversely alter the physiochemical environment to promote urate crystallization. Alteration of synovial lining permeability in OA may result in a circumstance where an effusion accumulates during the day, but in the evening when water is preferentially extruded over urate, synovial fluid urate levels increase disproportionately. Activation of synovial lining macrophages in response to urate crystals results in the generation of inflammatory mediators, particularly IL-1β, which may contribute to further OA cartilage degradation
Cartilage and Chondrocytes: Cartilage Damage, Urate Production, and Deposition
It is possible that chondrocytes themselves contribute to local accumulation of synovial fluid UA. Studies have demonstrated that chondrocytes in healthy cartilage are in a state of homeostatic autophagy, hypothesized to be necessary for maintaining normal cellular integrity, function, and survival [19]. However, chondrocytes in OA cartilage may become severely degenerated, with many of them undergoing programmed cell death (chondroptosis) [20]. As discussed further below, Shi et al. made the key observation that UA is a danger signal released by dying cells, which helps the immune system differentiate potentially deadly antigens from innocuous ones and has an adjuvant effect on the immune response [21]. Chondroptosis may therefore result in the release of UA which, in the right setting, could contribute to increased local UA concentrations, and in turn, lead to increased risk of MSU precipitation.
Regardless of the source of UA, some investigators have suggested that damaged cartilage may serve as a nidus for MSU crystal formation and/or deposition. In an assessment of the talar joints of 7855 adults, Muehleman et al. observed a strong correlation between the location of MSU crystal deposition and sites of OA articular cartilage lesions, occurring most commonly at sites where opposing articular surfaces were inadequately congruent with each other [22].
OA Synovium and Synovial Fluid: a Vehicle for Urate Accumulation and Precipitation?
OA synovial fluid perturbances have been implicated in promoting urate accumulation. For example, Simkin demonstrated that the synovium is more permeable to water than to UA, suggesting a mechanism for nocturnal rises in intra-articular UA concentrations. He proposed that OA synovial effusions, containing both water and UA, develop gradually during the day but resolve overnight when the joint is rested. Given the differences in the synovial permeability, water may be resorbed more rapidly than UA in the evening, concentrating UA in the joint space and promoting a circadian risk pattern for MSU crystallization [23]. This theory is consistent with the observation that acute gouty attacks tend to occur overnight. Physical trauma, which can result in an effusion independent of underlying arthritis, is known to promote gouty attacks and may also operate in part according to this mechanism [5].
In addition to fluctuations of UA concentrations in the OA joint, biomolecular characteristics unique to OA joint fluids may promote MSU crystallization by altering UA solubility. Degeneration of OA articular cartilage is accompanied by shedding of glycosaminoglycans and proteoglycans into the synovial fluid [24]. One of the major sulfated glycosaminoglycans in the matrix of joint tissues is chondroitin sulfate (CS), which can be further classified into chondroitin-6-sulfate (C6S), derived primarily from articular cartilage and related to the integrity of the articular surfaces, and chondroitin-4-sulfate (C4S), which is more widely distributed in the cartilage, synovium, ligaments, and menisci [25]. In comparing synovial fluid from healthy volunteers and patients afflicted with OA, Sharif et al. found that healthy volunteers had higher C6S levels and C6S/C4S ratios but lower C4S levels, compared to patients with OA [26]. Moreover, Uesaka et al. conducted a study correlating CS variables to severity of radiographic OA and noted that while early OA was associated with increases in the synovial fluid concentrations of all three chondroitins (C4S, C6S, and C6S/C4S ratio), advanced OA was characterized by decreases in both C6S concentrations and the C6S/C4S ratio, indicating a relative predominance of C4S [27].
The differences in absolute and relative amounts of C4S and C6S found in an OA joint based on radiographic degree of OA may be related to the zonal distribution of each of these isomers in the cartilage. According to Bayliss et al., C6S concentrations are highest in the surface layers of articular cartilage and decrease with progression toward the deeper mid-zone [28]. This finding suggests that while superficial cartilage degeneration in early OA may be reflected in increasing C6S and C6S/C4S ratios in synovial fluid, C4S may be preferentially shed once deeper cartilage layers are exposed.
Differences in synovial fluid CS profiles between healthy and OA joints have led investigators to ask whether CS concentrations may influence MSU crystallization. In vitro studies have demonstrated that the presence of CS may hasten MSU crystal formation, thus providing a possible explanation for why gout may have a predilection to target joints with underlying OA [29, 30]. In particular, CS4 has been shown to reduce UA solubility and enhance MSU crystallization [30]. It remains to be determined whether the ratio of C6S/C4S has any bearing on these findings, and whether these findings pertain in vivo. Moreover, while CS may affect UA crystal precipitation in patients with OA, at least one study suggests that it may also inhibit crystal-induced inflammation. In an in vitro study, Orlowsky et al. reported the ability of CS to attenuate MSU-induced production of IL-1β and tumor necrosis factor (TNF)-α by macrophages, suggesting a possible mechanism for the resolution of acute gouty inflammation [31].
UA and Gout as Possible Risk Factors for OA
In contrast to the paradigm that OA may provide an environment for enhanced MSU crystallization, other data suggest that gout may convey a risk for developing OA and that hyperuricemia itself may play one of several roles.
MSU Deposition: Mechanical and Biocellular Cartilage Alteration
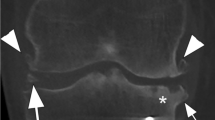
MSU deposits, in the form of tophi at or near articular surfaces, have long been appreciated to disrupt joint architecture in ways that can adversely affect joint mechanics and potentially trigger the pathophysiologic and cellular processes of OA (secondary OA) [32]. Sokoloff proposed that defects in the articular cartilage of a joint affected by OA facilitate tophus deposition, but that subsequently, tophi can erode into the surrounding tissues resulting in progressive structural damage [33]. Recent advances in imaging, particularly musculoskeletal ultrasound, have additionally led to the recognition of cartilage surface depositions of MSU crystals (“double contour sign”) in hyperuricemic subjects both with and without gout, including in joints that have not been affected by gouty attacks [34•]. The impact of these surface deposits on cartilage and their implications for OA are a matter of ongoing investigation (Fig. 2).

Musculoskeletal ultrasound illustrating two types of monosodium urate deposition. a Monosodium urate crystal deposition on the surface of articular cartilage. Shown is an ultrasound image of the first metatarsophalangeal (MTP) joint of a patient with gout, with the distal metatarsal head on the left. The heavy arrows point to a white (hyperechoic) line (double contour sign), indicating the presence of monosodium urate deposition on the surface of the articular cartilage. The articular cartilage itself appears as a dark (anechoic) zone below the white line, with the underlying bone surface beneath indicated by a second, hyperechoic white line. b Gout and OA in a single joint. Shown here is the first MTP joint of a different patient, again with the distal metatarsal head on the left. The heavy arrows point to a collection of tophaceous material around the joint, in a somewhat amorphous distribution. The thin arrows point to two hyperechoic white lines that represent bony cortex and appear to be discontinuous with the adjacent cortex. These discontinuities represent “step offs” that are the characteristic ultrasound appearance of osteoarthritis osteophytes
There is mounting data to suggest that MSU deposits may trigger cellular processes that compromise the structural integrity of cartilage. Liu et al. demonstrated that in vitro, MSU crystals induce matrix metalloproteinase (MMP)-3 and nitric oxide (NO) release from chondrocytes [35]. MMP-3 acts to break down non-helical, non-collagen domains of several cartilage matrix proteins [36]. NO can inhibit chondrocyte proteoglycan synthesis [37], impair chondrocyte viability [38], and enhance the catabolic activity of MMPs [39]. Both MMP-3 and NO release may promote cartilage damage. Others have shown that MSU crystals can directly reduce human chondrocyte viability and function [40••] and induce chondrocyte death [41].
MSU Triggers Inflammatory Cascades Implicated in OA
UA and MSU have garnered increasing attention in the past decade for their pivotal roles as triggers of inflammatory cascades. Martinon et al. made the seminal observation that MSU crystals activate the leukocyte NLRP3 inflammasome to drive IL-1β and IL-18 production [42]. IL-1β is considered a gatekeeper of inflammation and has pleotropic effects in multiple tissues, including those of the joint. In OA, IL-1β has been shown to promote cartilage degeneration by stimulating MMP secretion from chondrocytes, suppressing type II collagen and aggrecan expression and inducing the production of IL-6 and other catabolic cytokines and chemokines [43, 44]. IL-1β also participates in the induction of proinflammatory genes such as cyclooxygenase 2, TNF-α, and NO synthase. In addition to the effects noted earlier, NO generation can downregulate the endogenous IL-1β neutralizer, IL-1 receptor antagonist (IL-1Ra), potentially allowing IL-1β to promote its own efficacy [45] (Fig. 1).
The potential role of IL-1β as a mediator between gout and OA is supported by clinical studies examining the relationships between UA and inflammatory cytokines in synovial fluid and OA severity [46]. In a study of knee OA by Denoble et al., synovial fluid UA levels directly correlated with synovial fluid IL-18 and IL-1β levels, and the levels of all three molecules correlated with radiographic features of OA (osteophyte scores and joint space narrowing) [47••]. Taking these observations together, Denoble et al. suggested a cyclical model, in which chondrocyte death results in pericellular increases in UA concentration and facilitates microscopic MSU precipitation to promote more inflammation and neighbor chondrocyte death. Conceivably, the superimposition of actual gout or even simply increases in synovial fluid UA concentrations could accelerate or exacerbate these processes.
Potential Role of Soluble UA
Most studies of the adverse effects of gout focus on the role of crystals. However, soluble UA is itself a biologically active molecule, raising the possibility of a causal link between hyperuricemia and OA even in the absence of crystallization [48]. Mobasheri et al. reported that UA transporters are present on human articular chondrocytes, suggesting that these cells may internalize soluble UA with potential pro-oxidant effects [49, 50].
Using peripheral blood mononuclear cells harvested from patients with gout and healthy volunteers, Crişan et al. reported that leukocytes exposed to higher soluble UA concentrations demonstrate augmented cytokine generation in response to stimulation by toll-like receptor (TLR) ligands [51]. Of particular interest is the fact that the disequilibrium that favored inflammation was largely mediated by a significant downregulation of IL-1Ra. Whether these peripheral effects of soluble urate on leukocytes may impact OA in the joint, and whether soluble UA may have similar, direct effects on chondrocytes (or indirect effects through effects on synovial fibroblast-like and macrophage-like cells) remains to be determined.
Synovial Versus Serum UA
In contrast to synovial fluid levels, sUA is a readily accessible marker and therefore of greater potential utility to clinicians treating patients with OA. It is important, therefore, to ask whether sUA concentrations are relevant to OA profiling, either independently or through their influence on synovial UA levels. Several large retrospective studies have examined the relationship between sUA and OA at both the population and individual levels, but there have been conflicting results. Analysis of the National Health and Nutrition Examination Survey (NHANES) found no statistically significant relationships between sUA and the incidence, severity, and pathogenesis of OA [52]. Similarly, Mishra et al. examined serological parameters of oxidative stress (e.g., sUA, lipid panel, CRP) in control, OA, and rheumatoid arthritis (RA) subjects and found no significant differences in sUA levels among the three groups [53]. On the other hand, in a large epidemiological cross-sectional study, Acheson et al. found a significant association between sUA and OA [54]. Similarly, Sun et al. found a positive correlation between sUA and generalized OA among patients with hip OA but not among patients with knee OA [55]. This finding was replicated in the Ulm Osteoarthritis Study conducted by Gunther et al. involving 420 patients with hip OA and 398 patients with knee OA [56].
Some studies report conflicting associations of UA and OA, depending on whether serum or synovial fluid UA is examined, raising the question of whether there is a reliable correlation between serum and synovial fluid UA levels in various arthritides. Denoble et al. noted that in a cohort of patients with knee OA but no history of gout, serum UA concentrations were higher than synovial fluid UA concentrations in 85 % of subjects [47••]. They also demonstrated that the higher the sUA, the less reliable the correlation between serum and synovial fluid UA became [47••]. Furthermore, they ultimately found that only synovial fluid UA, and not sUA, was correlated significantly with OA severity, even though synovial fluid is considered to be an ultrafiltrate of plasma [47••].
However, other studies report a more straightforward relationship between serum and synovial UA. Wangkaew et al. conducted a comparative study of serum and synovial fluid UA levels in patients with gout and other arthritides, including OA, calcium pyrophosphate dehydrate (CPPD) disease, RA, septic arthritis, and ankylosing spondylitis. In each of these conditions, they reported a positive correlation between serum and synovial fluid urate levels; for OA in particular, the correlation coefficient was a striking 0.81. Intriguingly, OA patients had the second highest mean serum (6.1 mg/dl) and synovial fluid (5.9 mg/dl) UA levels after gout (6.7 and 7.1, respectively) [57]. In another study comparing the UA concentrations in serum and synovial fluid from patients with various joint disorders, paired serum and synovial fluid UA levels were not found to differ significantly in patients with OA or gout [58]. This finding held true even after adjusting for renal function and the use of medications that might influence UA concentrations [58]. However, the same study did find that UA levels were significantly higher in the serum than synovial fluid of patients with inflammatory arthritides such as RA and CPPD [58].
Therapeutic Implications
Despite the prevalence and impact of OA worldwide, no disease modifying anti-osteoarthritic drug (DMOAD) has received approval from the Food and Drug Administration [59]. Current treatments are mostly palliative and focused on analgesia, with joint replacement, if feasible, as the final option when palliation fails. Our growing insight into the role of inflammation in OA, and our nascent but evolving understanding of the possible role(s) for UA effects on OA cartilage, raise the possibility that targeting either sUA levels or MSU crystal-induced inflammation deserves consideration for OA trials. Moreover, even if sUA was shown only to reflect OA severity and/or progression without necessarily playing a pathogenic role, establishing sUA as a readily available biomarker to identify at-risk patients would greatly facilitate prospective OA drug trials.
Several small-scale studies have examined a potential role for colchicine, a standard anti-inflammatory in the gout treatment armamentarium, in the treatment of OA. Colchicine has been shown to suppress MSU-induced IL-1β release by the NLRP3 inflammasome [60•], suggesting a possible mechanism for colchicine impact on OA pathophysiology. Das et al. conducted a randomized controlled trial (RCT) of colchicine in 39 subjects with knee OA [61]. All subjects initially received piroxicam daily. If OA symptoms persisted despite 2 weeks of piroxicam, subjects were continued on piroxicam, received an intra-articular steroid injection, and were randomized to receive colchicine or placebo for 5 months. Compared to placebo, the addition of colchicine resulted in more durable relief of OA symptoms. Additionally, a significantly higher number of control patients reported pain in previously unaffected musculoskeletal areas as a significant new problem compared to the colchicine group, suggesting a possible role for colchicine in not only the treatment but also prevention of symptomatic OA in already predisposed individuals.
Aran et al. randomized 61 postmenopausal female patients with knee OA to receive either colchicine or placebo [62]. Both groups additionally received common OA treatments such as acetaminophen for rescue analgesia. Improvement in pain at the end of 3 months was significantly greater in the colchicine group, as assessed by both patients and physicians. Moreover, acetaminophen consumption was significantly lower in the colchicine group. One important difference between the studies conducted by Das and Aran was that in the former, 75 % of study participants demonstrated CPPD crystals, whereas in the latter, those who had evidence of CPPD disease on radiography were excluded from the study. Since colchicine is indicated for the treatment of CPPD disease (European League Against Rheumatism (EULAR) guidelines), the inclusion of CPPD patients in the Das study may have obfuscated OA-specific responses [63].
The primary data collection for a larger scale RCT of 120 patients with knee OA called colchicine effectiveness in symptom and inflammation modification in knee osteoarthritis (COLKOA) was completed in September 2015. In this study, 120 patients with knee OA were randomized to daily colchicine or placebo, in addition to their baseline analgesic regimen, and assessed for clinical response as well as levels of inflammatory markers after 16 weeks [59]. The study findings are still pending at this time.
The notion that directly lowering urate could reduce OA risk or burden is an intriguing one that has not yet been extensively evaluated in humans. In a mouse model of lung inflammation, Gasse et al. demonstrated that urate-lowering therapy could reduce IL-1β generation, suggesting that urate-lowering therapy may abrogate processes important to OA joint states [64]. Based on growing appreciation of metabolic syndrome as a risk factor for OA [65], Aibibula et al. employed a metabolic syndrome mouse model to show that mice fed a high fat diet experienced increased OA prevalence and severity, as well as increased activity of xanthine oxidase, the rate-limiting step in UA synthesis [66•]. The administration of the xanthine oxidase inhibitor febuxostat attenuated the histologic and radiographic changes of knee OA, even when the high fat diet was continued [66•]. Consistent with Gasse’s results, IL-1β levels tracked with the presence of OA, and were increased in response to the high fat diet and decreased in response to febuxostat use.
Conclusions
To date, no conclusive causal relationship between UA, gout, and OA has been established. However, there is mounting data to suggest that such a relationship might exist, though its directionality remains uncertain. Increasing understanding of the role of inflammation in OA development and progression, as well as the identification of MSU crystallization as an intercellular trigger of inflammatory responses, has promoted interest into the role of hyperuricemia and gout in OA pathogenesis. Advancements in musculoskeletal ultrasound may enable us to identify patients with asymptomatic hyperuricemia who may already have joint involvement in the absence of a diagnosis of gout and may be at risk for OA. Given the increasing worldwide burden of OA and the lack of DMOAD currently available, there is good justification for future studies to focus on understanding the relationship between UA and OA, and whether traditional gout medications could be repurposed for OA melioration.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Bruyere O et al. A consensus statement on the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO) algorithm for the management of knee osteoarthritis-From evidence-based medicine to the real-life setting. Semin Arthritis Rheum. 2016;45(4 Suppl):S3–11.
Vos T et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2163–96.
Kuo CF et al. Global epidemiology of gout: prevalence, incidence and risk factors. Nat Rev Rheumatol. 2015;11(11):649–62. Summarizes global epidemiology of gout in terms of prevalence and incidence in diverse regions of the world, as well as the evolving understanding of the associated risk factors for gout.
Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–41.
Roddy E, Doherty M. Gout and osteoarthritis: a pathogenetic link? Joint Bone Spine. 2012;79(5):425–7.
Fam AG, Stein J, Rubenstein J. Gouty arthritis in nodal osteoarthritis. J Rheumatol. 1996;23(4):684–9.
Lawrence RC et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778–99.
Lawrence RC et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II Arthritis Rheum. 2008;58(1):26–35.
Musumeci G et al. Osteoarthritis in the XXIst century: risk factors and behaviours that influence disease onset and progression. Int J Mol Sci. 2015;16(3):6093–112.
King KB, Rosenthal AK. The adverse effects of diabetes on osteoarthritis: update on clinical evidence and molecular mechanisms. Osteoarthr Cartil. 2015;23(6):841–50.
Sattui SE, Singh JA, Gaffo AL. Comorbidities in patients with crystal diseases and hyperuricemia. Rheum Dis Clin North Am. 2014;40(2):251–78.
Roddy E, Doherty M. Epidemiology of gout. Arthritis Res Ther. 2010;12(6):223.
Howard RG et al. Presence of gout is associated with increased prevalence and severity of knee osteoarthritis among older men: results of a pilot study. J Clin Rheumatol. 2015;21(2):63–71.
Kuo CF et al. Comorbidities in patients with gout prior to and following diagnosis: case-control study. Ann Rheum Dis. 2016;75(1):210–7.
Ding X et al. The associations of serum uric acid level and hyperuricemia with knee osteoarthritis. Rheumatol Int. 2016;36:567–73.
Roddy E, Zhang W, Doherty M. Gout and nodal osteoarthritis: a case-control study. Rheumatology (Oxford). 2008;47(5):732–3.
Kawenoki-Minc E et al. Osteoarthrosis and spondylosis in gouty patients. Analysis of 262 cases of gout. Reumatologia. 1974;12(3):267–7.
Roddy E, Zhang W, Doherty M. Are joints affected by gout also affected by osteoarthritis? Ann Rheum Dis. 2007;66(10):1374–7.
Carames B et al. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62(3):791–801.
Roach HI et al. Pathobiology of osteoarthritis: pathomechanisms and potential therapeutic targets. Curr Drug Targets. 2007;8(2):271–82.
Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425(6957):516–21.
Muehleman C et al. Association between crystals and cartilage degeneration in the ankle. J Rheumatol. 2008;35(6):1108–17.
Simkin PA. The pathogenesis of podagra. Ann Intern Med. 1977;86(2):230–3.
Inerot S et al. Articular-cartilage proteoglycans in aging and osteoarthritis. Biochem J. 1978;169(1):143–56.
Mourao PA. Distribution of chondroitin 4-sulfate and chondroitin 6-sulfate in human articular and growth cartilage. Arthritis Rheum. 1988;31(8):1028–33.
Sharif M et al. The relevance of chondroitin and keratan sulphate markers in normal and arthritic synovial fluid. Br J Rheumatol. 1996;35(10):951–7.
Uesaka S et al. Significance of chondroitin sulfate isomers in the synovial fluid of osteoarthritis patients. J Orthop Sci. 2002;7(2):232–7.
Bayliss MT et al. Sulfation of chondroitin sulfate in human articular cartilage. The effect of age, topographical position, and zone of cartilage on tissue composition. J Biol Chem. 1999;274(22):15892–900.
Burt HM, Dutt YC. Growth of monosodium urate monohydrate crystals: effect of cartilage and synovial fluid components on in vitro growth rates. Ann Rheum Dis. 1986;45(10):858–64.
Laurent TC. Solubility of sodium urate in the presence of chondroitin-4-sulphate. Nature. 1964;202:1334.
Orlowsky EW et al. Monosodium urate crystal induced macrophage inflammation is attenuated by chondroitin sulphate: pre-clinical model for gout prophylaxis? BMC Musculoskelet Disord. 2014;15:318.
Dalbeth N et al. Relationship between structural joint damage and urate deposition in gout: a plain radiography and dual-energy CT study. Ann Rheum Dis. 2015;74(6):1030–6.
Sokoloff L. The pathology of gout. Metabolism. 1957;6(3):230–43.
Gutierrez M et al. International Consensus for ultrasound lesions in gout: results of Delphi process and web-reliability exercise. Rheumatology (Oxford). 2015;54(10):1797–805. Develops first consensus-based definition for gout based on musculoskeletal ultrasound and demonstrates good reliability in a web-based exercise.
Liu R et al. Proline-rich tyrosine kinase 2 and Src kinase signaling transduce monosodium urate crystal-induced nitric oxide production and matrix metalloproteinase 3 expression in chondrocytes. Arthritis Rheum. 2004;50(1):247–58.
Goldring MB. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000;43(9):1916–26.
Taskiran D et al. Nitric oxide mediates suppression of cartilage proteoglycan synthesis by interleukin-1. Biochem Biophys Res Commun. 1994;200(1):142–8.
Blanco FJ et al. Chondrocyte apoptosis induced by nitric oxide. Am J Pathol. 1995;146(1):75–85.
Murrell GA, Jang D, Williams RJ. Nitric oxide activates metalloprotease enzymes in articular cartilage. Biochem Biophys Res Commun. 1995;206(1):15–21.
Chhana A et al. The effects of monosodium urate monohydrate crystals on chondrocyte viability and function: implications for development of cartilage damage in gout. J Rheumatol. 2013;40(12):2067–74. Investigates effect of MSU crystals on chondrocyte viability and function and provides a mechanism for which MSU crystals may result in cartilage damage.
Hwang HS et al. Monosodium urate crystal-induced chondrocyte death via autophagic process. Int J Mol Sci. 2015;16(12):29265–77.
Martinon F et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–41.
Gowen M et al. An interleukin 1 like factor stimulates bone resorption in vitro. Nature. 1983;306(5941):378–80.
Pretzel D et al. In vitro model for the analysis of synovial fibroblast-mediated degradation of intact cartilage. Arthritis Res Ther. 2009;11(1):R25.
Attur MG et al. Reversal of autocrine and paracrine effects of interleukin 1 (IL-1) in human arthritis by type II IL-1 decoy receptor. Potential for pharmacological intervention. J Biol Chem. 2000;275(51):40307–15.
Siebuhr AS et al. Inflammation (or synovitis)-driven osteoarthritis: an opportunity for personalizing prognosis and treatment? Scand J Rheumatol. 2016;45(2):87–98.
Denoble AE et al. Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proc Natl Acad Sci U S A. 2011;108(5):2088–93. Compares synovial fluid UA and inflammatory cytokine levels with knee OA severity, lending strong support for the role of the innate immune system in OA development and progression.
Nowatzky J et al. The role of uric acid and other crystals in osteoarthritis. Curr Rheumatol Rep. 2010;12(2):142–8.
Doblado M, Moley KH. Facilitative glucose transporter 9, a unique hexose and urate transporter. Am J Physiol Endocrinol Metab. 2009;297(4):E831–5.
Mobasheri A et al. Human articular chondrocytes express three facilitative glucose transporter isoforms: GLUT1, GLUT3 and GLUT9. Cell Biol Int. 2002;26(3):297–300.
Crisan TO et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann Rheum Dis. 2016;75(4):755–62.
Davis MA, Ettinger WH, Neuhaus JM. Obesity and osteoarthritis of the knee: evidence from the National Health and Nutrition Examination Survey (NHANES I). Semin Arthritis Rheum. 1990;20(3 Suppl 1):34–41.
Mishra R et al. A comparative analysis of serological parameters and oxidative stress in osteoarthritis and rheumatoid arthritis. Rheumatol Int. 2012;32(8):2377–82.
Acheson RM, Collart AB. New Haven survey of joint diseases. XVII. Relationship between some systemic characteristics and osteoarthrosis in a general population. Ann Rheum Dis. 1975;34(5):379–87.
Sun Y et al. Serum uric acid and patterns of radiographic osteoarthritis—the Ulm Osteoarthritis Study. Scand J Rheumatol. 2000;29(6):380–6.
Gunther KP et al. Clinical epidemiology of hip and knee joint arthroses: an overview of the results of the “Ulm Osteoarthrosis Study”. Z Rheumatol. 2002;61(3):244–9.
Wangkaew S et al. A comparative study of serum and synovial fluid levels of uric acid between patients with gout and other arthritides. J Med Assoc Thai. 2014;97(7):679–85.
Beutler AM et al. Soluble urate in sera and synovial fluids from patients with different joint disorders. Clin Exp Rheumatol. 1996;14(3):249–54.
Leung YY et al. Colchicine effectiveness in symptom and inflammation modification in knee osteoarthritis (COLKOA): study protocol for a randomized controlled trial. Trials. 2015;16:200.
Slobodnick A, Shah B, Pillinger MH, Krasnokutsky S. Colchicine: old and new. Am J Med. 2015;128(5):461–70. Summarizes pleotropic effects of colchicine and its potential utility in a number of different disease processes.
Das SK et al. A randomized controlled trial to evaluate the slow-acting symptom modifying effects of a regimen containing colchicine in a subset of patients with osteoarthritis of the knee. Osteoarthr Cartil. 2002;10(4):247–52.
Aran S, Malekzadeh S, Seifirad S. A double-blind randomized controlled trial appraising the symptom-modifying effects of colchicine on osteoarthritis of the knee. Clin Exp Rheumatol. 2011;29(3):513–8.
Zhang W et al. EULAR recommendations for calcium pyrophosphate deposition. Part II: management. Ann Rheum Dis. 2011;70(4):571–5.
Gasse P et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med. 2009;179(10):903–13.
Yoshimura N et al. Accumulation of metabolic risk factors such as overweight, hypertension, dyslipidaemia, and impaired glucose tolerance raises the risk of occurrence and progression of knee osteoarthritis: a 3-year follow-up of the ROAD study. Osteoarthr Cartil. 2012;20(11):1217–26.
Aibibula Z et al. Xanthine oxidoreductase activation is implicated in the onset of metabolic arthritis. Biochem Biophys Res Commun. 2016;472(1):26–32. Using a mouse model, this study provides rationale for potential use of direct urate lowering therapy to prevent cartilage degradation and inflammation commonly seen in metabolic syndrome.
Acknowledgments
The authors wish to thank Dr. Eric Morand, of this journal’s editorial board, for his helpful review of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
CY, MC, and AB declare that they have no conflict of interest. MHP reports grants from Takeda, Inc., personal fees from AstraZeneca, and personal fees from Crealta/Horizon, outside the submitted work. SK reports personal fees from Crealta/Horizon, outside the submitted work.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Crystal Arthritis
Michael H. Pillinger and Svetlana Krasnokutsky contributed equally to this work.
Rights and permissions
About this article

Cite this article
Yokose, C., Chen, M., Berhanu, A. et al. Gout and Osteoarthritis: Associations, Pathophysiology, and Therapeutic Implications. Curr Rheumatol Rep 18, 65 (2016). https://doi.org/10.1007/s11926-016-0613-9
Published:
DOI: https://doi.org/10.1007/s11926-016-0613-9