
Abstract
The idiopathic inflammatory myopathies (IIMs) comprise a group of autoimmune disorders that target skeletal muscle. They are characterized by typical laboratory and clinical features including muscle weakness, elevated muscle enzymes, characteristic histopathology of muscle biopsies, as well as electromyography abnormalities. The IIMs are divided into polymyositis, dermatomyositis, inclusion body myositis, nonspecific myositis, and immune-mediated necrotizing myopathy (IMNM). IMNM is distinguished by the absence of primary inflammation on muscle biopsy. IMNM may be associated with myositis-specific autoantibodies (i.e., anti-SRP and anti-HMGCR) and malignancy, in association with viral infections (HIV or hepatitis C), or in relation to other connective tissue diseases (i.e., scleroderma). Typical clinical findings such as severe muscle weakness, highly elevated creatine kinase (CK) levels, as well as resistance to conventional immunosuppressive therapy are associated with this subtype of IIM. This review provides an overview of this disease entity and focuses on its diagnosis and treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The idiopathic inflammatory myopathies (IIMs) comprise a group of autoimmune disorders that target skeletal muscle. Females are affected twice as often as males, and the incidence of IIM is estimated to be five to ten new cases/million population/year [1–4, 5••]. The IIMs are characterized by typical laboratory and clinical features including muscle weakness upon examination, elevation of muscle enzymes, characteristic histopathology of muscle biopsies, as well as typical electromyography (EMG) abnormalities including irritable myopathy and insertional irritability. In the case of dermatomyositis, typical rashes are noted including heliotrope rash and Gottron’s papules [1]. While Bohan and Peter criteria [6] have been recognized as the predominant diagnostic criteria for IIM, other criteria have been proposed. There has been a lack, however, of uniformity in the updated diagnostic criteria proposed which can pose a challenge when conducting studies in IIM [1, 6–9]. One of the significant criticisms of existing classification criteria was the lack of recognition of immune-mediated necrotizing myopathy (IMNM) as a distinct subgroup of IIM [10••]. Under some existing classification schemes, for example the Bohan and Peter Criteria [6], IMNM might be classified as polymyositis. According to the diagnostic and classification criteria proposed by the Muscle Study Group/European Neuro Muscular Centre (MSG/ENMC) in 2004, the IIMs are divided into polymyositis (PM), dermatomyositis (DM), inclusion body myositis (IBM), nonspecific myositis, and immune-mediated necrotizing myopathy [4]. IMNM is distinguished from the other groups by the scant presence of an inflammatory infiltrate on muscle biopsy [5]. However, the diagnosis of IMNM cannot be made on muscle biopsy alone, and with the identification of myositis-specific antibodies, as well as further identification of other histologic features on muscle biopsy other than necrosis alone, it is recognized that IMNM is not one, but at least several different diseases [11] . Furthermore, muscle cell necrosis is a nonspecific feature that can occur in many muscle-damaging processes, and IMNM must be differentiated from toxic myopathies (i.e., drug-induced myopathies), endocrinopathies, trauma-induced myopathies [12], and muscular dystrophies [13].
Subtypes of AINM
IMNM may be associated with several different causes, including malignancy [5••, 10••, 14–20, 21••, 22, 23], viral infections such as hepatitis C [24] and HIV [25–27] [10••] [28–31], connective tissue diseases such as scleroderma [5••, 20, 23, 32••, 33–35], and subsets of patients associated with different myositis-specific antibodies including autoantibodies against signal recognition particle (anti-SRP) [36–39],or against 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), especially in statin-exposed patients [11, 23, 32••].
In this paper, studies regarding the diagnosis and treatment of IMNM are discussed. Common clinical features and investigations used in the diagnosis of IMNM, as well as treatment strategies used will be reviewed.
Diagnosis of IMNM
Classification of IMNM
As previously stated, not all classification criteria for IIM take into account IMNM. For example, under the Bohan and Peter criteria, muscle biopsy findings are not used to differentiate between PM, DM, and IMNM. At the time this classification was developed, it was recognized that some myositis patients had necrosis on biopsy with a lack of inflammation, but this was thought to be accounted for by patients with cancer-associated myositis [13].
In 1991, Love et al. [40] proposed a new approach to the classification of idiopathic inflammatory myopathy that utilized myositis-specific autoantibodies, including anti-SRP, to divide patients into homogeneous groups. The majority of the seven anti-SRP patients analyzed were African-American females. The presentation of myositis in these patients was severe and included myalgias, distal weakness, falls, atrophy of muscles, mechanic’s hands, and palpitations. However, these criteria did not address IMNM as a distinct entity.
In 1997, Targoff et al. [7] proposed the addition of myositis-related autoantibodies to the classification scheme for IIM, one of the antibodies being anti-SRP. However, the proposed muscle biopsy criteria did not specifically mention necrosis, and there was no distinct subset of IMNM.
Troyanov et al. [9] developed a clinic-serologic classification utilizing myositis-associated and myositis-specific antibodies that also included overlap features, with one of these myositis-specific antibodies being anti-SRP. According to this classification, subsets of IIM are divided into PM, DM, and overlap myositis (OM) that include features such as polyarthritis, Raynaud’s and interstitial lung disease among others, and cancer-associated myositis. Once again, IMNM is not considered as a distinct subset, and this criterion does not take histopathological findings into account. Fernandez et al. [41] undertook a study involving a retrospective cohort of 178 patients with clinic-pathologic features suggestive of IIM, in order to determine the pathologic features of each serologic subset of IIM and to propose guidelines to diagnose IIM according to both clinic-serologic and pathologic classifications. Necrotizing myopathy was considered a distinct pathological subset of IIM, characterized as having features of numerous necrotic-regenerative fibers with absent or minimal inflammatory changes, with absent or focal MHC-1 overexpression, on muscle biopsy. When compared to the clinic-serologic classification of Troyanov, most cases of IMNM would have been classified as OM or PM.
In 2003, a classification scheme was proposed that was greatly based on muscle biopsy findings [4]. According to this classification scheme, the IIMs were divided into DM, PM, inclusion body myositis (IBM), and nonspecific myositis. In addition, IMNM was recognized as a distinct form of autoimmune muscle disease in which muscle biopsies reveal muscle cell necrosis and degeneration with a lack of significant inflammatory infiltrates. Under this classification criteria, a patient is classified as having IMNM if he or she has onset of myopathy over 18 years of age, a sub-acute or insidious onset of muscle weakness, proximal greater than distal and neck flexor greater than neck extensor weakness, and elevated creatine kinase (CK) levels. Furthermore, patients need to fulfill one out of the following three laboratory criteria: irritable myopathy on EMG, magnetic resonance imaging (MRI) with STIR of muscle tissue revealing diffuse or patchy increased signal, or myositis-specific antibodies detected in the serum. In addition, the muscle biopsy findings are key and must show many necrotic muscle fibers as the predominant abnormal histological feature.
Classification and diagnostic criteria for IMNM is an evolving field, due to the heterogeneity of the etiology of this entity. It has been recognized that IMNM is not one specific disease, but is composed of at least several different diseases, and that data in the form of clinical trials, for example, is needed to determine subsets of IMNM based on autoantibodies, medication exposure, and specific muscle biopsy features [13].
Clinical Features of IMNM
It has been recognized in many studies that patients with IMNM present with a common phenotype that includes profound weakness, highly elevated CK levels, myalgias, and resistance to conventional immunosuppressive medications [14, 42]. These features may prompt clinicians to consider IMNM as a potential diagnosis. Different subsets of disease may have unique and/or overlapping presentations. In some clinical situations, other symptoms and signs such as dysphagia, Raynaud’s phenomenon, and lung involvement may be observed. While interstitial lung disease (ILD) appears to be uncommon, malignancy may be seen in association with autoantibody-related IMNM or as a stand-alone entity.
Bronner at al. [14] reported on a group of eight patients who presented with symmetrical proximal muscle weakness with a sub-acute onset, CK more than 10 times elevated, and muscle biopsy showing widespread atrophy, necrosis, and regeneration with absence of inflammatory cell infiltrates. The myopathy was steroid-responsive. Distal weakness and dysphagia were also noted. However, myositis-specific antibody testing for anti-SRP was not done.
When IMNM is suspected based on clinical presentation and biopsy, autoantibodies can be ordered (e.g., anti-SRP, anti-HMGCR). Cohorts of these subsets have also been studied for typical clinical findings at diagnosis. Anti-SRP antibody can now be screened with commercially available immunoblots [22], and HMGCR testing is now commercially available through the RDL reference laboratory [43].
SRP antibodies are quite specific for myopathy and have been quoted as accounting for 5 % of cases of inflammatory myopathies [11]. The approximate incidence of anti-SRP antibodies in patients with IMNM is 15 % [39]. However, anti-SRP antibodies have also been found in patients with limb-girdle muscular dystrophy type 2A and other diseases [44]. Patients with IMNM associated with anti-SRP antibodies typically present with the phenotype previously described of extreme weakness, myalgias, high CK values, irritable myopathy on EMG, inflammation on MRI muscle imaging, and profound necrosis with absence of inflammation on muscle biopsy. Raynaud’s is a variable feature. Table 1 summarizes some typical clinical findings noted in studies involving patients with anti-SRP-related IMNM.
HMG-CoA reductase (HMGCR) is the pharmacologic target of the statins. Christopher-Stine and colleagues performed screening for novel autoantibodies among a group of patients with necrotizing myopathy on biopsy without a known underlying etiology and identified a subgroup of these patients who had autoantibodies recognizing 200- and 100-kD proteins; the 100-kD protein was later identified as HMG-CoA reductase (HMGCR), the pharmacologic target of the statins, and the 200-kD protein as an HMGCR dimer [23, 31]. Mammen et al. [45••] reported that 6 % of patients in the Johns Hopkins Myositis Center have anti-HMGCR antibodies, and based on an incidence of autoimmune myopathy of four per 100,000 per year, it is therefore estimated that the incidence of anti-HMGCR myopathy is roughly two per million per year [46]. Patients with HMGCR-associated myopathy present with weakness, although not always as dramatic as seen with SRP myopathy, myalgias, high CK levels, irritable myopathy on EMG, inflammation on MRI muscle imaging, and predominant necrosis on muscle biopsy with the absence of inflammation. Not all patients were statin-exposed, but among those who were 50 years old and above, 92.3 % had been on a statin [43]. Overall, statin-naïve patients were found to be younger, with higher CK levels at presentation, non-white and less responsive to treatment [32••].
The following clinical findings can be used to help make a diagnosis of IMNM in the proper clinical setting. For the purpose of this review, the focus is on anti-SRP-associated IMNM, anti-HMGCR-associated IMNM, and paraneoplastic IMNM.
Weakness
Anti-SRP-Associated IMNM
Severe weakness, at presentation and throughout the course of illness, has been observed in anti-SRP-associated IMNM. Therefore, in a patient presenting with severe weakness, mainly proximal but also some distal, a diagnosis of IMNM should be considered. This observation has been made in several studies worldwide.
It has been reported that there is a predominance of female and African-American patients in this patient population [11, 61]. In addition, it is our clinical experience that these patients tend to be younger than patients with another cause of IMNM; for example, in the study by Miller et al. [36], the mean age of onset of SRP myopathy was 48, which is younger than other cohorts (i.e., statin-associated cohorts) [62].
In the United States, Miller at al. [37] analyzed a group of seven patients with myopathy and serum anti-SRP antibodies, ranging in age from 32 to 70 years. These patients presented with severe proximal muscle weakness that rapidly developed over a period of months. Kao et al. [38] identified 16 anti-SRP-positive patients with polymyositis and found that this group of patients presented with early, severe proximal muscle weakness and atrophy compared to the control groups of patients with PM that were negative for the SRP autoantibody. In Canada, Troyanov et al. [9] studied a cohort of 100 French-Canadian patients and identified two patients with anti-SRP autoantibodies; both had sudden disease onset, and one presented with severe muscle weakness causing an extrathoracic restrictive breathing syndrome.
Hengstman et al. [39] carried out a retrospective systematic assessment of the clinical, laboratory and histological characteristics of 23 anti-SRP-positive patients from six European centers and compared data to a large group of SRP-negative myositis patients. Anti-SRP-positive patients presented with severe symmetrical proximal muscle weakness that resulted in marked disability. Weakness was rapidly progressive; patients had difficulty walking and standing; and the weakness progressed over a matter of months.
Similar findings have been noted in Asia. In Japan, Takada et al. [47] studied 21 anti-SRP-positive patients with myositis. Muscle weakness was always symmetrical and mostly in proximal muscles. Sixty percent of patients had severe muscle weakness at initial evaluation that then rapidly progressed from time of onset to severe disability. A retrospective study by Sugie et al. [48] retrospectively analyzed seven patients with anti-SRP-related myopathy; all patients had a sub-acute onset of symptoms, and one patient presented with rapid development of severe muscle weakness.
Wang et al. [49] studied a cohort of Chinese patients with anti-SRP antibodies. Of 16 patients studied, 14 patients presented with chronic progression of proximal limb weakness. A study by Zheng et al. identified 12 patients with SRP myopathy whose main symptoms were proximal limb weaknesses [50].
A recent case series of an Australian cohort of myositis patients confirmed five patients with histologically confirmed myositis who had anti-SRP antibodies [51]. Four patients had severe, rapidly progressing proximal muscle weakness, peaking within 4–8 weeks of onset of symptoms in three patients.
While the weakness observed in studies is routinely proximal muscle weakness, some studies have found distal weakness in SRP-positive patients as well [40]. Love et al. reported on a group of seven anti-SRP-positive patients who presented with weakness including distal weakness and a history of falls. Bulbar involvement has also been reported; from Japan, a recent observational study reported on a subset of patients with IMNM who were positive for the SRP autoantibody, and these patients exhibited severe limbs weakness and atrophy as well as bulbar and trunk muscle involvement [52••]. A recent case series from Japan involving 100 patients with inflammatory myopathy with anti-SRP antibody reported on severe neurological symptoms including profound limb, trunk, and bulbar muscle weakness with atrophy [45••].
Weakness can be sudden and severe. A review of autoimmune necrotizing myopathies by Allenbach et al. [53] described the onset of SRP-related IMNM as sudden in onset, unlike most other inflammatory myopathies that seem to have a more insidious onset. Myalgias are seen in 66–80 % of cases, and over 50 % of patients present severe proximal muscular impairment (i.e., muscular strength inferior or equal to 3/5 on the Medical Research Council scale). In fact, just weeks after onset of symptoms, some patients may be so weak as to be severely bed-ridden or wheelchair-bound.
Anti-HMGCR-Associated IMNM
Weakness is also a common clinical feature of anti-HMGCR antibody-associated IMNM. Watanabe et al. [52••] recently published an observational study involving eight subjects with HMGCR antibody-associated IMNM. Symmetrical and proximal limb weakness was observed, with both arms and legs equally affected; however, weakness was found to be mild, with severe limb weakness of grade ≤2/5 assessed by manual muscle strength (Medical Research Council scale grade) not being observed. This was in contrast to SRP antibody myositis patients studied, who exhibited severe muscle weakness.
While Grable-Esposito et al. did not directly test for anti-HMGCR autoantibodies, they did observe a cohort of 25 patients that developed a necrotizing myopathy while on statins and progressive symptoms after stopping statins. All patients had symmetric, proximal leg and arm weakness, and distal weakness also developed in five patients [20]. In the study by Christopher-Stine et al. [23], all 16 patients with HMGCR antibody-associated IMNM developed acute or sub-acute onset of proximal muscle weakness. Interestingly, the level of CK elevation did not necessarily correspond with the degree of muscle weakness noted, since some patients had very high CK levels with only mild weakness upon examination. Again, this seems to be in contrast to anti-SRP-associated IMNM, where severe weakness is often observed.
Ramanathan et al. examined a cohort of six Australian patients with statin exposure who developed an HMGCR antibody-associated IMNM; all patients presented with painless proximal weakness following statin therapy, which persisted after statin cessation [54]. Weakness was of varying severity.
Severe weakness has been noted in this patient population, however. Allenbach et al. [21••] recently described a cohort of 45 European patients with anti-HMGCR antibodies and IMNM. Statin exposure was recorded in 44.4 % of patients. Almost all patients had weakness (97.7 %), frequently severe (Medical Research Council [MRC] 5 ≤ 3 in 75.5 %). Most of the cases (64.4 %) had a sub-acute onset of weakness (<6 months), although three patients (6.6 %) had a slowly progressive course over more than 10 years. In most patients, elevation of CK level correlated with muscle strength. In addition, a significant correlation was found between titres of the anti-HMGCR antibodies and the MRC score of the weakest muscle groups. A similar correlation between titers and muscle strength was noted by Werner et al. among HMGCR antibody-related IMNM patients [55].
Dysphagia
Anti-SRP-Associated IMNM
Dysphagia has been noted in anti-SRP-positive patients with IMNM. Miller et al. [37] noted three out of the seven patients in their study experienced dysphagia. In the European study by Hengstman et al. [39], patients with anti-SRP-associated myopathy had a higher rate of dysphagia than SRP-negative patients. Hanisch et al. [56] reported a case of a patient with anti-SRP-associated myopathy who had a severe presentation of dysphagia along with dysarthrophonia, bilateral facial palsy, and loss of reflexes, as well as paresis of both proximal and distal muscles, although it was suspected that he had an overlap with another neurological syndrome. In the Australian cohort previously mentioned [51], three patients had dysphagia. In the Suzuki’s study [45••], 41 patients presented with dysphagia. Similarly, in the study by Takada et al. [47], three out of 21 patients showed signs of dysphagia.
A recent critical review that focussed on the classification, diagnosis, and management of IIMs [57] found that patients with SRP myopathy can have dysphagia as a clinical feature of their disease. In the review by Allenbach et al. [53], dysphagia has been reported in 30–69 % of cases.
Anti-HMGCR-Associated IMNM
Dysphagia has been noted in patients with anti-HMGCR-associated IMNM. Grable-Esposito et al. [20] observed three patients with dysphagia in their study of 25 patients with likely IMNM in the setting of statin use, although HMGCR autoantibodies were not specifically tested for. Christopher-Stine et al. [23] noted dysphagia in ten out of 16 (63 %) of patients with HMGCR antibody-related IMNM. Allenbach et al. noted dysphagia in 26.7 % (12/45) of patients with HMGCR-related IMNM [21••].
CK Levels
Anti-SRP-Associated IMNM
Patients with anti-SRP-associated IMNM present with highly elevated CK levels. The magnitude of the rise in CK levels can help reinforce the diagnosis of IMNM.
In the study by Miller et al. [37], serum CK levels were very high (3000 to 25,000 IU/L). Takada et al. [47] observed that patients with SRP myopathy had highly elevated serum CK levels as well, ranging from 1387 to 9900 IU/L (mean ± SD 5016 ± 2452 IU/L). In Suzuki’s case series of 100 patients, mean CK levels were 6161 ± 4725 IU/L [45••], and Wang et al. [49] also noted highly elevated CK levels (from 400 to 9082 IU/L). This finding was reinforced in the review by Allenbach et al. [53] commented on high CK levels in this subgroup of patients (range between 6600 and 15 000 IU/L). The retrospective study by Sugie et al. [48] was also characterized by patients with very high CK levels (2000–15,000 IU/L).
Anti-HMGCR-Associated IMNM
As seen in anti-SRP-associated myopathy, CK levels tend to be elevated with anti-HMGCR-associated IMNM as well. Mammen et al. noted that CK levels are higher than what one would normally associate with IIM; however, many patients did not exhibit any myositis symptoms until CK levels were in excess of several thousands of international units per liter [42]. In the study by Grable-Esposito et al. [20], patients with statin-associated necrotizing myopathy had highly elevated serum CK levels (mean 8203 IU/L, range 3000 to 17,280 IU/L), and Christopher-Stine et al. [23] also noted high CK levels in their study of anti-HMGCR-associated patients with IMNM (mean maximum CK of 10,333 IU/L). Watanabe et al. similarly observed high CK levels in this patient population that varied from 3028 to 10,452 IU/L [52••], and Ramanathan et al. noted serum CK levels that ranged from 2700 to 16,200 IU/L [54].
Although CK values can reach very high levels, a variation in values may be seen. For example, Mammen et al. [32••] analyzed 45 patients who were HMGCR-positive and found CK levels from between 200–35,000 IU/L. The CK value may also correspond with muscle strength. Allenbach et al. noted mean CK levels of 6941 ± 8802 IU/L which correlated with muscle strength evaluated by manual muscle testing [21••].
EMG
EMG findings in IMNM are typically of an irritable myopathy and include spontaneous fibrillation potentials, positive sharp waves, and insertional irritability. These findings are seen in the majority of patients with IMNM.
Anti-SRP-Associated IMNM
Miller et al. noted classic myopathic EMG findings including features of muscle membrane irritability (i.e., spontaneous fibrillation potentials and positive sharp waves) and small amplitude, polyphasic, brief motor unit potentials in all patients tested [37]. In the Takada’s paper [47], EMG findings in proximal muscles included myopathic motor unit potentials and prominent spontaneous activity, again found in all patients tested.
Anti-HMGCR-Associated IMNM
EMG findings are typical of an irritable myopathy. Grable-Esposito et al. [20] noted abnormal spontaneous activity in the form of fibrillation potentials, positive sharp waves, and myotonic or pseudomyotonic discharges as the most frequent findings seen on EMG. Christopher-Stine et al. [23] and Mammen et al. [32••] also noted irritable myopathy on EMG in anti-HMGCR-positive patients; in the study by Christopher-Stine et al., irritable myopathy was found in 88 % of patients tested. EMG findings of irritable myopathy (low amplitude, short duration motor unit potentials) were similarly seen in the study by Watanabe et al. [52••].
MRI
Anti-SRP-Associated IMNM
Zheng et al. [50] undertook a study to evaluate muscle MRI changes and the role of MRI in the monitoring of therapy in 12 patients with anti-SRP-associated myopathy. MRI imaging revealed thigh muscle edema and fatty infiltration in all 12 patients. CK elevation did not relate to the degree of edema seen on imaging. Sugie et al. [48] reported on findings of diffuse inflammation and edema seen on skeletal muscle MRI, most prominent in the proximal muscles of all four extremities.
Anti-HMGCR-Associated IMNM
Christopher-Stine et al. noted evidence of muscle edema on bilateral thigh MRI in anti-HMGCR patients with IMNM [23]. Watnabe et al. also noted focal or diffuse abnormal signaling in the trunk and limb muscles on MRI in a similar patient population [52••].
Muscle Biopsy
Anti-SRP-Associated IMNM
The classic features on muscle biopsy for IMNM include widespread necrosis with lack of inflammatory infiltrates. However, muscle tissue necrosis can be a nonspecific finding seen in other conditions that affect muscle, for example, in the setting of endocrinopathies, or from damage due to toxic exposures [13]. Certain features of muscle histology may help distinguish IMNM from other causes of necrotizing myopathy. Upregulation of the MHC class I antigen in non-necrotic muscle fibers can be used to help identify those who have IMNM. Moreover, complement deposition on capillaries and on the sarcolemma of non-necrotic fibers is more characteristic of an immune-mediated process in patients with a necrotizing muscle biopsy [58]. Regarding anti-SRP-associated IMNM vs anti-HMGCR IMNM, there are some characteristic features (i.e., capillary involvement) that can help distinguish between the two, as described below. Anti-SRP-Associated IMNM:
Characteristic features of IMNM have been noted on muscle biopsy from patients with anti-SRP antibody-mediated myopathy. These features include necrosis, degeneration and regeneration of muscle fibers, and lack of inflammatory infiltrates. Capillary involvement has also been observed. Both Kao [38] and Hengstman [39] observed necrotic and regenerating muscle fibers on biopsies from patients with anti-SRP-associated myopathy. In addition, Hengstman et al. commented on the presence of swollen capillaries in 85 % of muscle biopsy specimens. Miller et al. [37] also noted atrophic and hypertrophic fibers as well as C5b-9 membrane attack complex deposits on muscle fiber surfaces and capillaries. Of note, the presence of hypertrophic fibers can also be seen with dystrophies, so immune-histochemical stains for the dystrophies should be performed to exclude these conditions [27]. Regarding capillary involvement, Miller et al. also observed a reduction in the capillary density (capillary index) and enlargement of endomysial capillaries on biopsy as a characteristic feature of anti-SRP-associated IMNM.
From Japan, similar to other studies, Suzuki et al. [45••] commented on necrotizing myopathy on muscle biopsy. Takada et al. examined muscle biopsy specimens from 11 patients with anti-SRP-associated myositis. All 11 patients demonstrated muscle fiber necrosis and/or regeneration, but only one had infiltration of inflammatory cells on biopsy [47]. Kawabata et al. described a case of a pediatric patient from Japan with anti-SRP-associated myopathy whose biopsy revealed active necrotic and regenerating fibers, with mild inflammatory changes [59]. In fact, inflammatory changes may be observed in IMNM. For example, in their review paper, Allenbach et al. [53] noted the total absence of muscular inflammation seen on biopsy as a distinguishing feature of anti-SRP IMNM; however, they also noted that minor endomysial inflammation may be observed. The study by Wang et al. [49] examined the muscle biopsies of 16 patients with anti-SRP myopathy; these biopsies exhibited features of necrotic and regenerating fibers in all 16 patients, lymphocytic infiltration in 11, and features of muscular dystrophy in seven patients. Nine patients showed deposition of membrane attack complexes in necrotic muscle fibers, two with such deposition around capillaries and 11 with focal or diffuse MCH Class I expression in sarcolemma or cytoplasm of muscle fibers.
Anti-HMGCR-Associated IMNM
Muscle biopsy specimens from patients with anti-HMGCR-associated IMNM reveal necrosis, enhanced MHC-I expression on necrotic and non-necrotic muscle fibers, abnormal capillary morphology, and lack of inflammatory infiltrate. One study revealed that 94 % of patients with anti-HMGCR autoantibodies had muscle biopsy specimens showing prominent myofiber necrosis with minimal inflammation [23]. Further immuno-histochemical studies showed membrane attack complex on the small blood vessels on the surface of non-necrotic myofibers, abnormal capillary morphology, and expression of class I MHC on the surface of non-necrotic myofibers. Grable-Esposito et al. [20] also reported on MHC-I upregulation on both necrotic and non-necrotic muscle fibers. Muscle biopsy features from the study by Allenbach et al. primarily exhibited necrosis and/or muscle fiber regeneration, muscle fiber size irregularity and presence of atrophic fibers, lack of inflammatory infiltration, C5b-9 deposition predominantly on necrotic fibers and occasionally muscle capillaries, and overexpression of MHC class I on regenerative or necrotic fibers [21••]. Watanabe et al. noted regenerating muscle fibers clearly stained by polyclonal anti-HMGCR antibodies on immune-histochemistry staining [52••]. Ramanathan et al. [54] also commented on a pauci-immune necrotizing myopathy seen on biopsy.
Other Clinical Findings
Other clinical features have been noted in patients with both anti-SRP- and anti-HMGCR-associated IMNM.
Anti-SRP-Associated IMNM
Raynaud’s Phenomenon
Love et al. identified Raynaud’s phenomenon as a symptom seen in their cohort of anti-SRP-associated myositis patients [40], and Takada et al. [47] reported on one out of 23 anti-SRP-positive myositis patients with Raynaud’s. Kao et al. [37] noted Raynaud’s in 76 % of their patients studied. The overall association of Raynaud’s phenomenon with anti-SRP-associated myopathy is quoted as 20–26 % in the recent review by Allenbach et al. [53].
Interstitial Lung Disease (ILD)
Interstitial lung disease (ILD) may also be noted in anti-SRP-associated myositis patients. Kao et al. [38] reported the finding of ILD in three anti-SRP-positive patients who actually did not have myositis; however, these patients had connective tissue disease or another autoantibody associated with ILD. In the European, multicenter retrospective assessment published by Hengstman et al. [39], ILD was reported in patients with anti-SRP-associated myopathy, found in 25 % of the patients studied. ILD was also reported in one patient in the study by Takada as previously mentioned [47]. Sugie et al. [48] reported on one patient positive for anti-SRP autoantibody as well as Jo-1 autoantibody who exhibited symptoms and signs of severe ILD; other patients with anti-SRP autoantibodies only presented with mild ILD.
Myalgias
Myalgia has been noted as a clinical feature of anti-SRP-associated myopathy [60]. In their review of IMNM, Allenbach et al. noted that myalgia is seen in 66–80 % of cases of IMNM [53].
Cardiac
Targoff et al. [7] reported on four out of 12 anti-SRP-associated myopathy patients with cardiac involvement; three had arrhythmias, and one had cardiac fibrosis. Love et al. [40] noted palpitations among the anti-SRP-associated myopathy patients reported in their study. Abnormalities in heart rate and rhythms have been noted in other studies; Allenbach et al. [53] noted in their review paper abnormal electrocardiogram findings in 50 % of cases, including arrhythmias or nonspecific conduction defects, with clinical signs of cardiac involvement present in less than 20 % of cases. In the study by Hengstman [39], transthoracic echocardiography demonstrated abnormalities in over half of cases, although for 50 % of these patients were related to previous coronary disease. However, other studies did not observe an increased incidence of cardiac events in patients with anti-SRP-associated IMNM [38].
Anti-HMGCR-Associated IMNM
Raynaud’s Phenomenon
Raynaud’s phenomenon is not a frequently seen feature of anti-HMGCR-associated IMNM. Allenbach et al. [21••] noted that Raynaud’s was reported in seven out of 45 patients in their review. Christopher-Stine et al. [23] reported only two of 16 (13 %) patients had Raynaud’s phenomenon.
ILD
ILD does not appear to be a phenotypic feature of HMGCR-associated IMNM; higher rates overall have not been seen [21••, 23, 32••].
Myalgias
Myalgias were observed in many studies. Studies reported 50–75 % of patients reporting myalgias [21] [23].
Paraneoplastic IMNM
Malignancy and myositis has been an observed association in previous studies, predominantly seen in association with DM, and this relationship has also been noted in cases of PM. However, myopathy with little or no inflammation but predominant muscle fiber necrosis has also been noted with malignancy alone [15], in the absence of PM or DM. There are no systematic studies assessing the overall risk of cancer in patients with IMNM. In a case series by Bronner et al. [14] of patients with IMNM, three out of eight patients had cancer preceding the diagnosis of myopathy or following it within 3 years of diagnosis.
Amato et al. commented that the most common associated malignancies seen with IMNM are gastrointestinal tract adenocarcinomas and small cell and non-small cell carcinomas of the lung, and recommended that a full malignancy workup should be performed on all patients who present with IMNM [5••]. Similar observations were noted by Dalakas et al. in their review of IIMs [22], as well as Levin et al. [17]. Due to the paucity of cases reported, standardized incidence ratios have not been calculated [61].
Malignancy may also been seen in the context of anti-HMGCR-associated IMNM. The study by Christopher-Stine et al. noted 13 % of patients had a malignancy [23], and in the study of Allenbach et al. [21••], five out of 45 patients had cancer.
The authors recommended a full malignancy workup at the time of diagnosis of IMNM, similar to one that would be undertaken at the time of diagnosis of PM or DM.
Treatment of IMNM
A variety of immunosuppressant medications have been used in the treatment of IMNM. No controlled trials of immunosuppressive therapy in IMNM exist, although the use of such medications has been documented in several reports. It is the authors’ opinion that IMNM often requires aggressive, sustained immunosuppression and may be resistant to treatment. Relapses can occur with tapering of immunosuppression and with re-exposure to statins in the case of anti-HMGCR-associated myopathy. Whether some anti-HMGCR-positive patients with significant cardiovascular risk factors can ever be safely re-exposed to statins remains to be determined.
Immunosuppressants that have been used in the treatment of IMNM include methotrexate, azathioprine, mycophenolate mofetil, rituximab, and often IVIG [11]. Combination therapy is often utilized. No optimum treatment strategy has yet been defined for this disease.
Anti-SRP-Associated IMNM
In the treatment of anti-SRP-associated myopathy, prednisone and another agent such as methotrexate, azathioprine, IVIG, or rituximab are often needed, and long-term immunotherapy is often required. Despite treatment, residual deficits are often seen [45••]. Miller et al. [37] noted six out of seven patients who were partially or totally resistant to steroid treatment. Kao et al. [38] reported that ten out of 16 patients required treatment by at least three different drugs and there was often a need for combined immunosuppressive therapy (methotrexate, azathioprine, cyclosporine, cyclophosphamide, tacrolimus, and infliximab) or IVIG. Favorable treatment results were seen in one third of patients. Hengstman et al. [39] noted that most patients were treated with a combination of corticosteroids and immunosuppressive medications, and that only two patients were treated with prednisone alone. Other treatments used were methotrexate (n = 15 patients), azathioprine (n = 11), cyclosporine (n = 5), IVIG (n = 5), cyclophosphamide (n = 2), and plasmapheresis (n = 2). Most patients were unable to be weaned off medications, and the relapse rate on tapering of dosages or stoppage of drugs was 70 %. Most patients had persisting residual muscle weakness despite treatment. In the investigation by Wang et al. [49], all patients required the addition of methotrexate or IVIG, and relapses were common during steroid tapering.
In the study by Suzuki et al. [45••],in addition to prednisone, 62 (77 %) of 81 patients required additional immunotherapy, including IVIG (n = 33), intravenous methylprednisolone plus therapy (n = 32), tacrolimus (n = 22), methotrexate (n = 11), azathioprine (n = 11), cyclosporine (n = 9), intravenous cyclophosphamide (n = 3), or plasma exchange (n = 3). Over half of the patients were refractory to various treatment regimens, and despite a decrease of the patients’ CK levels, recovery of muscle weakness was incomplete. Most patients required long-term immunosuppressive therapy and experienced side effects.
Rituximab has been studied in the treatment of other types of myopathy, including PM, DM and juvenile DM in several case series [62–64].
Rituximab therapy has been shown to result in improved symptoms and reduced steroid use as well as reduction of SRP antibody levels in refractory SRP-associated cases [65]. In the series by Valilyl et al., six of eight anti-SRP patients, previously refractory to other immunosuppressive therapy, were treated with rituximab and demonstrated improvement in muscle strength as well as a decline in CK levels as early as 2 months after treatment. These patients were able to reduce their doses of adjunctive steroids as well as maintain response for 12–18 months post initial dosing. However, a study involving two patients with anti-SRP myopathy treated with rituximab failed to demonstrate a dramatic clinical improvement [66]. Conversely, another study involving two patients with anti-SRP myopathy who were treated with steroids, plasma exchange, and rituximab therapy resulted in partial to complete, sustained recovery of muscle strength in both patients [67].
Maeshima et al. [60] reported a case of refractory anti-SRP-associated myopathy that had failed treatment with methotrexate, tacrolimus, and infliximab who ultimately responded to abatacept, with reduction of previously elevated CK levels back to normal.
Anti-HMGCR-Associated IMNM
A review of the literature does indicate that most patients require immunosuppressive drugs and/or immunomodulatory treatment, in the form of prednisone and another agent, most commonly methotrexate or IVIG. These treatments have resulted in improvement in strength and lowering of CK values [20, 21••, 23, 68].
In the case of anti-HMGCR-associated IMNM, Ramanathan et al. [54] reported on six patients treated with high-dose steroids and subsequently with varying regimens of other immunosuppressive agents, including IVIG (n = 5), plasmapheresis (n = 2), and additional therapy including: methotrexate (n = 6),cyclophosphamide (n = 2),rituximab (n = 2),azathioprine (n = 1), or cyclosporine (n = 1). Despite therapy, five out of six patients experienced a relapse, with between zero and three relapses per patient after a mean of 4.5 years of follow-up (range 1.5–11 years). Relapses were associated with steroid tapering or cessation, and most patients experienced ongoing myopathic symptoms for periods ranging from 6 months to 11 years after cessation, despite institution of immunosuppressive therapy.
Christopher-Stine et al. [23] noted that most anti-HMGCR-positive patients had a very modest initial response to prednisone and required combination immunosuppressive therapy. Rituximab and IVIG were helpful additions to prednisone and azathioprine or methotrexate. Similarly, Werner et al. [55] noted that prednisone and two other immunosuppressive agents were needed at some time during the treatment course of both statin-exposed and statin-naïve HMGCR-positive patients with IMNM. Allenbach et al. [21••] reported that all but two anti-HMGCR-positive patients with IMNM were treated with steroids as first-line treatment, frequently associated with the use of additional immunosuppressive drugs including methotrexate (n = 20) and cyclophosphamide (n = 1) or intravenous immunoglobulin (n = 10). One severely weak patient was also treated by plasmapheresis as first-line treatment. All patients but one who were treated for more than 1 year required intensification by DMARD in the form of additional DMARD treatment. The mean duration of treatment for 39 treated patients was 34.1 ± 40.8 months (range 1–180 months), and by the end of the study, no patient had been able to stop their treatment for a prolonged time over 1 year because of relapse.
Interestingly, as it was previously noted, refractory anti-SRP patients usually respond well to treatment with rituximab, whereas statin-naïve anti-HMGCR patients may not (12). It has also been suggested that anti-HMGCR-positive patients, statin-exposed, often improve dramatically with IVIG, whereas statin-naïve anti-HMGCR patients may be refractory to any immunosuppressive therapy [13]. This reiterates the point that IMNM is a heterogeneous diagnosis, and that patients with anti-SRP-associated IMNM are different from those with anti-HMGCR-associated IMNM, despite similarities in presentation and investigations.
Paraneoplastic IMNM
Wegener et al. [16] published a case report and review of the literature regarding the treatment of paraneoplastic IMNM. Treatment of the underlying cancer is the first step in therapy; however, additional treatment in the form of steroids, IVIG, or other immunosuppressive drugs may also be warranted.
Conclusions
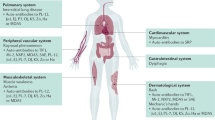
IMNM may be associated with several underlying etiologies. Patients will often present with high CK levels and a sub-acute to acute, rapid, severe onset of weakness. IMNM may be associated with myositis-specific antibodies, in the context of statin use, or with malignancy with or without known autoantibodies. A lack of extra-muscular manifestations is a general rule but other clinical findings may be noted. Other causes of necrosis of muscle tissue should be ruled out, such as toxin or drug-induced, or secondary to trauma or infection. Certain clinical features, including proximal muscle weakness, elevated CK levels, and necrosis on biopsy can lead to additional investigations and workup for IMNM (see Figure 1). IMNM often needs aggressive, prolonged immunosuppression, but there are no large trials addressing this issue and only a small numbers of patients have been studied.

Diagnostic algorithm for IMNM
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Cox S, Limaye V, Hill C, Blumbergs P, Roberts-Thomson P. Idiopathic inflammatory myopathies: diagnostic criteria, classification and epidemiological features. Int. J. Rheum. Dis. 2010:117–24.
Iago PF, Albert SO, Andreu FC, Xavier MG, Jose RP, Jordi PL, et al. Pregnancy in adult-onset idiopathic inflammatory myopathy: report from a cohort of myositis patients from a single center. Semin. Arthritis Rheum. [Internet]. 2014; Available from: http://www.ncbi.nlm.nih.gov/pubmed/24906908.
Gazeley DJ, Cronin ME. Diagnosis and treatment of the idiopathic inflammatory myopathies. Ther. Adv. Musculoskelet. Dis. [Internet]. 2011;3:315–24. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3383495&tool=pmcentrez&rendertype=abstract.
Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord England. 2004:337–45.
Amato A a, Greenberg S a. Inflammatory myopathies. Continuum (Minneap. Minn). [Internet]. 2013;19:1615–33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24305450. An excellent overview of inflammatory myopathies with helpful author commentary.
Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344–7.
Targoff IN, Miller FW, Medsger Jr TA, Oddis CV. Classification criteria for the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 1997;9:527–35.
Tanimoto K, Nakano K, Kano S, Mori S, Ueki H, Nishitani H, et al. Classification criteria for polymyositis and dermatomyositis. J Rheumatol. 1995;22:668–74.
Troyanov Y, Targoff IN, Tremblay J-L, Goulet J-R, Raymond Y, Senécal J-L. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore). 2005;84:231–49.
Lazarou IN, Guerne P-A. Classification, diagnosis, and management of idiopathic inflammatory myopathies. J. Rheumatol. [Internet]. 2013;40:550–64. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23504386. A comprehensive review of IIMs, including detailed analysis of current advances in classification, diagnosis and management.
Quinn C, Salameh JS, Smith T, Souayah N. Necrotizing myopathies: an update. J Clin Neuromuscul Dis USA. 2015;16:131–40.
Castro C, Gourley M. Diagnosis and treatment of inflammatory myopathy: issues and management. Ther. Adv. Musculoskelet. Dis [Internet]. 2012;4:111–20. Available from: http://tab.sagepub.com/content/early/2011/12/19/1759720X11425092.abstract.
Mammen AL. Necrotizing myopathies: beyond statins. Curr Opin Rheumatol. 2014;26:679–83.
Bronner IM, Hoogendijk JE, Wintzen AR, Van Der Meulen MFG, Linssen WHJP, Wokke JHJ, et al. Necrotising myopathy, an unusual presentation of a steroid-responsive myopathy. J Neurol. 2003;250:480–5.
Smith B. Skeletal muscle necrosis associated with cainoma. J Pathol England. 1969;97:207–10.
Wegener S, Bremer J, Komminoth P, Jung HH, Weller M. Paraneoplastic necrotizing myopathy with a mild inflammatory component: a case report and review of the literature. Case Rep. Oncol. 2010:88–92.
Levin MI, Mozaffar T, Al-Lozi MT, Pestronk A. Paraneoplastic necrotizing myopathy: clinical and pathological features. Neurology. 1998:764–7.
Sampson JB, Smith SM, Smith AG, Singleton JR, Chin S, Pestronk A, et al. Paraneoplastic myopathy: response to intravenous immunoglobulin. Neuromuscul Disord. 2007;17:404–8.
Galani E, Bonakis A, Christodoulou C, Klouvas G, Angeliki D, Skarlos D. Can cetuximab affect paraneoplastic myopathy? J Neurooncol. 2009;93:437–8.
Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve. 2010;41:185–90.
Allenbach Y, Drouot L, Rigolet A, Charuel JL, Jouen F, Romero NB, et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statin. Medicine (Baltimore). [Internet]. 2014;93:150–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24797170. A comprehensive analysis of anti-HMGCR positive patients in a European cohort.
Dalakas MC. Review: an update on inflammatory and autoimmune myopathies. Neuropathol. Appl. Neurobiol. 2011:226–42.
Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010;62:2757–66.
Satoh J, Eguchi Y, Narukiyo T, Mizuta T, Kobayashi O, Kawai M, et al. Necrotizing myopathy in a patient with chronic hepatitis C virus infection: a case report and a review of the literature. Intern Med [Internet]. 2000;39:176–81. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10732841.
Wrzolek MA, Sher JH, Kozlowski PB, Rao C. Skeletal muscle pathology in AIDS: an autopsy study. Muscle Nerve USA. 1990;13:508–15.
Snider WD, Simpson DM, Nielsen S, Gold JW, Metroka CE, Posner JB. Neurological complications of acquired immune deficiency syndrome: analysis of 50 patients. Ann Neurol. 1983;14:403–18.
Liang C, Needham M. Necrotizing autoimmune myopathy. Curr Opin Rheumatol. 2011;23:612–9.
Stern R, Gold J, DiCarlo EF. Myopathy complicating the acquired immune deficiency syndrome. Muscle Nerve. 1987:318–22.
Simpson DM. Myopathy associated with human immunodeficiency virus (HIV) but not with zidovudine. Ann Intern Med USA. 1988:842.
Simpson DM, Bender AN. Human immunodeficiency virus-associated myopathy: analysis of 11 patients. Ann Neurol. 1988;24:79–84.
Lange DJ, Britton CB, Younger DS, Hays AP. The neuromuscular manifestations of human immunodeficiency virus infections. Arch Neurol. 1988;45:1084–8.
Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme a reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63:713–21. Landmark paper regarding initial identification of the HMGCR autoantibody in a cohort of patients with IMNM.
Ellis E, Tan JA, Lester S, Tucker G, Blumbergs P, Roberts-Thomson P, et al. Necrotizing myopathy: clinicoserologic associations. Muscle Nerve. 2012;45:189–94.
Bhansing KJ, Lammens M, Knaapen HK, van Riel PL, van Engelen BG, Vonk MC. Scleroderma-polymyositis overlap syndrome versus idiopathic polymyositis and systemic sclerosis: a descriptive study on clinical features and myopathology. Arthritis Res. Ther. [Internet]. 2014;16:R111. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4060195&tool=pmcentrez&rendertype=abstract
Ranque B, Authier F-J, Le-Guern V, Pagnoux C, Berezne A, Allanore Y, et al. A descriptive and prognostic study of systemic sclerosis-associated myopathies. Ann Rheum Dis. 2009;68:1474–7.
Reeves WH, Nigam SK, Blobel G. Human autoantibodies reactive with the signal-recognition particle. Proc Natl Acad Sci USA. 1986;83:9507–11.
Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J. Neurol. Neurosurg. Psychiatry. 2002:420–8.
Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004;50:209–15.
Hengstman GJD, ter Laak HJ, Vree Egberts WTM, Lundberg IE, Moutsopoulos HM, Vencovsky J, et al. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Ann Rheum Dis England. 2006;65:1635–8.
Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991;70:360–74.
Fernandez C, Bardin N, De Paula AM, Salort-Campana E, Benyamine A, Franques J, et al. Correlation of clinicoserologic and pathologic classifications of inflammatory myopathies: study of 178 cases and guidelines for diagnosis. Medicine (Baltimore). [Internet]. 2013;92:15–24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23269233
Mammen AL, Pak K, Williams EK, Brisson D, Coresh J, Selvin E, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res. 2012;64:269–72.
Albayda J, Mammen AL. Is statin-induced myositis part of the polymyositis disease spectrum? Curr. Rheumatol. Rep USA. 2014;16:433.
Amato AA, Barohn RJ. Evaluation and treatment of inflammatory myopathies. J Neurol Neurosurg Psychiatr England. 2009;80:1060–8.
Suzuki S, Nishikawa A, Kuwana M, Nishimura H, Watanabe Y, Nakahara J, et al. Inflammatory myopathy with anti-signal recognition particle antibodies: case series of 100 patients. Orphanet J Rare Dis England. 2015;10:61. Comprehensive case series analayzing a large number of patients with anti-SRP associated myopathy.
Mohassel P, Mammen AL. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle Nerve. 2013:477–83.
Takada T, Hirakata M, Suwa A, Kaneko Y, Kuwana M, Ishihara T, et al. Clinical and histopathological features of myopathies in Japanese patients with anti-SRP autoantibodies. Mod Rheumatol Japan. 2009;19:156–64.
Sugie K, Eura N, Kobayashi Y, Sawa N, Ueno S. Clinicopathological and neuroradiological features of myopathy associated with antibodies to signal recognition particle (SRP). Eur. J. Intern. Med. [Internet]. Elsevier; 2015;24:e120. Available from: doi:10.1016/j.ejim.2013.08.306
Wang L, Liu L, Hao H, Gao F, Liu X, Wang Z, et al. Myopathy with anti-signal recognition particle antibodies: clinical and histopathological features in Chinese patients. Neuromuscul Disord. 2014;24:335–41.
Zheng Y, Liu L, Wang L, Xiao J, Wang Z, Lv H, et al. Magnetic resonance imaging changes of thigh muscles in myopathy with antibodies to signal recognition particle. Rheumatology (Oxford)England. 2015;54:1017–24.
Basnayake SK, Blumbergs P, Tan JA, Roberts-Thompson PJ, Limaye V. Inflammatory myopathy with anti-SRP antibodies: case series of a South Australian cohort. Clin Rheumatol Germany. 2015;34:603–8.
Watanabe Y, Suzuki S, Nishimura H, Murata K, Kurashige T, Ikawa M, et al. Statins and myotoxic effects associated with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase autoantibodies: an observational study in Japan. Medicine (Baltimore) USA. 2015;94:e416. Helpful paper analyzing anti-HMGCR associated IMNM in a Japanese cohort.
Allenbach Y, Benveniste O. Acquired necrotizing myopathies. Curr. Opin. Neurol. [Internet]. 2013;26:554–60. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23995277
Ramanathan S, Langguth D, Hardy TA, Garg N, Bundell C, Rojana-Udomsart A, et al. Clinical course and treatment of anti-HMGCR antibody-associated necrotizing autoimmune myopathy. Neurol Neuroimmunol neuroinflammation USA. 2015;2:e96.
Werner JL, Christopher-Stine L, Ghazarian SR, Pak KS, Kus JE, Daya NR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum USA. 2012;64:4087–93.
Hanisch F, Müller T, Stoltenburg G, Zierz S. Unusual manifestations in two cases of necrotizing myopathy associated with SRP-antibodies. Clin Neurol Neurosurg. 2012;114:1104–6.
Tansley S, Gunawardena H. The evolving spectrum of polymyositis and dermatomyositis-moving towards clinicoserological syndromes: a critical review. Clin Rev Allergy Immunol. 2013;1–10.
Preuße C, Goebel HH, Held J, Wengert O, Scheibe F, Irlbacher K, et al. Immune-mediated necrotizing myopathy is characterized by a specific Th1-M1 polarized immune profile. Am J Pathol. 2012;181:2161–71.
Kawabata T, Komaki H, Saito T, Saito Y, Nakagawa E, Sugai K, et al. A pediatric patient with myopathy associated with antibodies to a signal recognition particle. Brain Dev. 2012;34:877–80.
Maeshima K, Kiyonaga Y, Imada C, Iwakura M, Hamasaki H, Haranaka M, et al. Successful treatment of refractory anti-signal recognition particle myopathy using abatacept. Rheumatol. (United Kingdom). 2014:379–80.
Naert E, De Bleecker JL, Lumen N, Rottey S. Necrotizing myopathy as a paraneoplastic syndrome associated with renal cell carcinoma. Acta Clin Belg England. 2015;70:61–4.
Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601–7.
Sultan SM, Ng KP, Edwards JC, Isenberg DA, Cambridge G, Edwards JCW. Clinical outcome following B cell depletion therapy in eight patients with refractory idiopathic inflammatory myopathy. Clin Exp Rheumatol. 2008;26:887–93.
Lambotte O, Kotb R, Maigne G, Blanc FX, Goujard C, Delfraissy JF. Efficacy of rituximab in refractory polymyositis. J Rheumatol. 2005;32:1369–70.
Valiyil R, Casciola-Rosen L, Hong G, Mammen A, Christopher-Stine L. Rituximab therapy for myopathy associated with anti-signal recognition particle antibodies: a case series. Arthritis Care Res. 2010;62:1328–34.
Whelan BR, Isenberg DA. Poor response of anti-SRP-positive idiopathic immune myositis to B-cell depletion. Rheumatology (Oxford) England. 2009:594–5.
Arlet J-B, Dimitri D, Pagnoux C, Boyer O, Maisonobe T, Authier F-J, et al. Marked efficacy of a therapeutic strategy associating prednisone and plasma exchange followed by rituximab in two patients with refractory myopathy associated with antibodies to the signal recognition particle (SRP). Neuromuscul Disord England. 2006;16:334–6.
Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord. 2007;17:194–200.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Christopher-Stine reports personal fees from Walgreens, Novartis, Idera Pharmaceuticals, Ono Pharma UK, Marathon Pharmaceuticals, MedImmune/Astra Zeneca, and Mallinckrodt. In addition, Dr. Christopher-Stine has a patent HMGCR assay with royalties paid to Inova Diagnostics.
Dr. Basharat reports personal fees as a round-table participant for Novartis and Amgen.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Inflammatory Muscle Disease
Rights and permissions
About this article

Cite this article
Basharat, P., Christopher-Stine, L. Immune-Mediated Necrotizing Myopathy: Update on Diagnosis and Management. Curr Rheumatol Rep 17, 72 (2015). https://doi.org/10.1007/s11926-015-0548-6
Published:
DOI: https://doi.org/10.1007/s11926-015-0548-6