
Abstract
The idiopathic inflammatory myopathies are a heterogeneous group of disorders affecting both adults and children. Clinical features can include muscle weakness, skin disease and internal organ involvement. A large number of autoantibodies, directed against cytoplasmic or nuclear components, can now be identified in these patients and specific clinic-serological syndromes have been described. Laboratory testing to identify many of these autoantibodies is becoming easier, and here we discuss the clinical utility of autoantibodies in myositis both in terms of facilitating diagnosis, predicting disease course and informing management decisions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
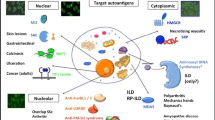
Autoantibodies directed against intracellular antigens form part of the diagnosis in many rheumatological conditions. They can be detected in approximately 80 % of adults and 60 % of children with idiopathic inflammatory myopathies (IIM) and many are very disease-specific; that is, they are not found in healthy individuals or other rheumatological disorders. The prevalence of these ‘myositis specific autoantibodies’ (MSA) varies with age at disease onset and in some cases the population studied [1]. The specificity of many autoantibodies means they provide a useful means to confirm a suspected diagnosis of IIM, although it is noteworthy that their absence does not preclude this diagnosis. ‘Myositis-associated autoantibodies’ (MAA) are typically identified in patients with overlap syndromes, and these patients may or may not have muscle disease as a dominant feature. The term MSA can in some cases be considered a misnomer, as patients may have minimal or no muscle disease: Dermatomyositis sine myositis or amyopathic myositis is reported to account for 20–30 % adult cases but is rare in juvenile disease [2–5]. In addition, the most well-described autoantibody-derived myositis sub-group, the anti-synthetase syndrome, can be considered as a spectrum where patients with anti-Jo-1 autoantibodies commonly present with more diffuse disease with significant muscle involvement, whilst those with anti-PL12, anti-KS and anti-OJ are more likely to have disease restricted to the lung and may never develop muscle involvement [6]. The term MSA will continue to be used throughout this review for convenience.
Here, we discuss the utility of myositis-specific and -associated autoantibodies in disease diagnosis and adopting a stratified approach to further investigation and management.
Diagnosis
IIM is an umbrella term encompassing a heterogeneous group of conditions. The feature of dermatomyositis distinguishing it from other IIM subtypes is the characteristic rash; however, similarly to the variability of muscle involvement, the pathognomic rash of DM can be subtle and easily missed and, particularly in juvenile disease, can be atypical. This can make the distinction between the classic sub-groups of dermatomyositis and polymyositis difficult. The rarity of IIM, combined with the potentially subtle examination findings and the general heterogenicity of these diseases, can lead to diagnostic delay. For inclusion body myositis (IBM) mean diagnostic delays of 4–5 years have been reported [7, 8].
The use of appropriate classification criteria and the division of IIM into appropriate subgroups is important both clinically and for future research study design. Work to establish an international consensus for diagnostic criteria in IIM is ongoing [9]. The Bohan and Peter criteria, although widely used, have long been considered outdated and fail to consider the utility of autoantibody testing or other potentially useful diagnostic techniques such as MRI. Autoantibody testing can be an invaluable tool to assist with diagnosis in a patient with symptoms consistent with IIM, in addition to other patient groups such as those patients with ‘idiopathic’ interstitial lung disease where an underlying connective tissue disease needs to be excluded. The new classification criteria for adult and juvenile IIM currently in development includes anti-Jo-1 positivity as a variable in its model and, in the absence of muscle biopsy, this confers the highest score in predicting IIM [9]. We believe that other anti-synthetase and mysotis-specific autoantibodies should be considered to have a similar diagnostic ‘weight’, but unfortunately as yet testing for these autoantibodies is not always readily available.
Anti-Jo-1 autoantibody testing is widely available and anti-Jo-1 is the most common autoantibody identified in adult patients with IIM but is rare in juvenile disease. A significant limitation of the clinical utility of autoantibodies in patients with myositis, particularly those with juvenile-onset disease, has been the availability of testing. Standard immunological techniques such as indirect immunofluorescence are of limited value, and a negative antinuclear antibody (ANA) does not preclude the presence of autoantibodies in patients with myositis, many of which produce a cytoplasmic staining pattern. There is a growing interest in simple, non-specialised laboratory techniques to detect other autoantibodies in myositis, and recent publications have reflected this with several recent papers describing techniques such as ELISA and laser bead immunoassays to detect different MSA, including those commonly found in juvenile disease [10–14].
Management
Predicting Disease Course
MSA are typically mutually exclusive, and the identification of autoantibodies and subsequent serological patient sub-classification can provide more detailed prognostic information than the broader clinically defined entities such as polymyositis and dermatomyositis. The disease phenotype associated with many of the autoantibodies that can be identified in patients with IIM is now well established, and has been the subject of recent reviews [15•, 16] (Table 1),
Myositis Syndromes
Anti-Synthetase Syndrome
The antisynthetase syndrome is well described and clinical features classically include myositis, interstitial lung disease, Raynaud’s phenommenon, mechanic’s hands, Gottron’s lesions, arthritis and pyrexia. Anti-Jo-1 is the most common MSA identifiable in adult patients with myositis, and is found in approximately 30 % of cases. The remaining antisynthetase autoantibodies (PL12, PL7, OJ, EJ, KS, Zo, Ha) can each be found in <5 %. While all are associated with the antisynthetase syndrome features listed above, disease presentation and the prevalence of pulmonary manifestations does vary depending on the specific associated autoantibody. While muscle disease is common in patients with anti-Jo1, anti-PL-7 or anti-EJ, patients with anti-PL-12, anti-KS or anti-OJ in contrast often have predominant ILD [16]. Of 166 Japanese patients with anti-synthetase antibodies, 29 % anti-Jo-1, 16 % anti-EJ, 10 % anti-PL-7 and 11 % anti-PL-12 presented initially with ILD alone, and although many subsequently developed myositis and other disease features, this was less likely in those with anti-PL-12, anti-KS and anti-OJ. In contrast, in those patients with myositis alone at presentation, nearly all subsequently developed ILD during follow-up [17]. In this way, the anti-synthetase syndrome can be thought of as a disease spectrum, and those patients with early pulmonary involvement or predominant respiratory symptoms may initially present to the respiratory clinic. Anti-synthetase antibodies were retrospectively identified in 6.6 % of 198 patients diagnosed with idiopathic interstitial pneumonia and, crucially, of those found to have an anti-synthetase antibody, just under 50 % had no extra-pulmonary features [18]. These patients are important to recognise, as a diagnosis of CTD-associated rather than idiopathic ILD is likely to influence both treatment and prognosis. Isolated ILD without myositis in patients with anti-synthetase syndrome is a risk factor for poor survival [19•].
The most common cause of death in this group is pulmonary fibrosis, and patients with non-Jo-1 antisynthetase antibodies have been found to have a worse survival than those with anti- Jo-1 [20•, 6]. Non-Jo-1 patients have also been found to have a greater delay in diagnosis compared to Jo-1 patients [20•]. This may reflect differences in disease presentation but also the reduced availability of testing for ‘non-standard’ autoantibodies. As diagnostic delay may be a contributory factor to the prognostic difference seen, the potential clinical benefit of wider MSA testing is clearly evident.
Anti-synthetase autoantibodies are rare in juvenile-onset disease and are found in <5 %. Where they have been identified, patients appear to have a similar disease phenotype to adults and are at risk of developing ILD. This is an important complication and, in a recent study of mortality in juvenile-onset IIM, while the number of deaths were small, ILD was the most common cause of death and anti-synthetase autoantibodies were associated with an increased risk of mortality [21].
Clinically Amyopathic Myositis
The term clinically amyopathic myositis (CADM) or myositis sine myositis is used to describe those patients who present with dermatological features of DM without any muscle involvement clinically or histologically. The prognosis in patients presenting in this way can vary widely, but MSA identification can assist clinicians in monitoring for anticipated complications. For example, patients with anti-SAE have been described in UK, European and Japanese cohorts [22–25]. These patients typically present with CADM first, and then progress to myositis with a higher frequency of dysphagia and gastrointestinal disease [25]. In contrast, patients with anti-MDA5 autoantibodies also commonly present with minimal or no muscle involvement; however, this autoantibody is associated with ILD in predominantly Caucasian cohorts and rapidly-progressive ILD with a high mortality in East Asian cohorts [26–28].
Amyopathic myositis is recognised in children but it is rare, and more often patients have mild or progressive muscle disease [29, 30]. Anti-MDA5 and anti-SAE autoantibodies have both been described in juvenile-onset disease. Anti-SAE is rare and, while the clinical phenotype has yet to be defined, disease presentation appears similar to that described in adults (personal data). Anti-MDA5 in contrast is relatively common in juvenile-onset disease and has been found in 38 % of Japanese cohorts and 7.4 % of UK juvenile patients [31, 32]. This is an important MSA to identify as, similarly to adults, there appears to be an association with ILD in UK children and rapidly-progressive ILD in Japanese children [31, 32].
Necrotising Autoimmune Myositis
Necrotising autoimmune myopathy is a recently recognised subgroup of IIM characterised by marked myofibre necrosis with minimal or no inflammatory infiltrate on muscle biopsy. Patients typically present with acute proximal weakness and a very high creatinine kinase level [33]. This pattern can also be seen as a result of drug or toxin exposure. Occasionally, patients may present atypically with an insidious onset and may be misdiagnosed as muscular dystrophy. The detection of anti-SRP or anti-HMGCR autoantibodies both facilitates diagnosis and identifies those patients likely to respond to immunomodulatory drugs. Patients with anti-SRP autoantibodies may present with features such as dysphagia, cardiac muscle involvement and arthritis in addition to muscle disease. Furthermore, this group of patients may be refractory to standard myositis treatment regimens and require a more aggressive treatment approach. Anti-HMGCR autoantibodies are strongly associated with necrotising autoimmune myopathy and also statin use, although it is noteworthy that only 40–60 % patients have previously been exposed to statins [12, 34].
Inclusion Body Myositis
Although classified as an IIM, inclusion body myositis has several features distinguishing it from other inflammatory myopathies, including a characteristic pattern of weakness with slowly progressive muscle weakness and atrophy. It is the most common acquired muscle disease in those over 50 years and, unlike other IIM and connective tissue diseases in general, is more common in men. Muscle biopsy typically reveals both inflammatory and degenerative features, with characteristic findings including rimmed vacuoles and amyloid deposits that are absent in other IIM subtypes. While both disease presentation and muscle histology are classically distinctive, diagnosis is difficult as the disease is rare, presentation can be atypical, and the sensitivity of muscle biopsy at presentation can be poor, as many of the pathological features do not develop until later [35, 36].
The relatively recent discovery of an autoantibody associated with IBM, anti-Mupp44, which targets cytosolic 5’-Nucleotidase 1A, is an exciting finding [37–40]. Further studies have shown that this autoantibody has a high sensitivity and specificity for IBM, suggesting it would be a useful tool in identifying this patient group [39, 40, 41•]. Anti-Mupp44 autoantibodies provide an important opportunity to identify patients with IBM at an earlier stage, preventing unnecessary treatment with immunosuppression, to which they would not be expected to respond, in addition to repeated invasive tests such as muscle biopsy in order to reach the diagnosis.
Overlap Syndromes
Muscle inflammation can also occur in other connective tissue diseases, including Scleroderma, SLE and Sjogrens syndrome in addition to Rheumatoid Arthritis, so-called myositis overlap syndromes. These are not common, and a recent analysis of overlap syndromes in a Brazilian myositis cohort identified overlap syndromes in 14 % of patients [42]. In this context, the identification of MAA or MSA is useful and can alert the clinician to an increased risk of muscle involvement in addition to other disease features in patients presenting with features of connective tissue disease. For example, in Scleroderma, patients with anti-PmScl autoantibodies often have a phenotype similar to the anti-synthetase syndrome. They are more likely to have limited cutaneous disease, muscle disease, ILD and calcinosis compared to anti-PmScl-negative patients. They are less likely to develop pulmonary arterial hypertension and have a better overall survival [43].
While MAA including anti-PmScl, anti-U1-RNP and anti-Ku are typically associated with myositis-overlap, so-called MSA have also been identified in this patient group, further blurring the boundaries between these two terms and reflecting challenges in disease classification. Furthermore, whilst MSA are typically mutually exclusive, other MAA such as anti-Ro52 can co-exist and may modify the disease phenotype. Anti-Ro52 has been identified in up to 56 % of those with anti-Jo-1 autoantibodies [44–46], and in some studies has been associated with more severe ILD [47, 48]. A more recent study failed to confirm this but did show patients with anti-Ro52 autoantibodies in addition to anti-Jo-1 had more severe myositis, joint impairment, an increased incidence of malignancy and reduced survival [49]. It is important to note that the identification of a MAA such as anti-Ro52 does not preclude the presence of an MSA, the latter being more informative with regard to disease phenotype.
Important Disease Complications
Malignancy
Malignancy represents a major cause of mortality in adult patients with myositis and there is a clearly established association between DM and the development of malignancy. The increased risk is higher in patients with DM than PM, and overall there is a threefold increase in risk of malignant disease for all cancer subtypes after diagnosis of DM [50, 51]. Standard clinical practice after making a diagnosis of IIM in an adult patient would be to conduct a ‘malignancy screen’, although there is little guidance as to how extensive screening should be. MSA can be useful in identifying those patients who may be more likely to develop a malignancy and in whom screening should therefore be more thorough. Whilst no specific cancer sub-type is associated with DM, anti-TIF1γ and anti-NXP2 autoantibodies have both been associated with an increased risk of malignancy in adult patients [52, 53]. It is intriguing that these autoantibodies are the most commonly identified in juvenile populations where no association with malignancy exists [54, 55].
Interstitial Lung Disease
ILD is a leading cause of mortality in both adult and juvenile-onset disease. As described above, it is associated with the anti-synthetase syndrome, anti-MDA5 autoantibodies, and overlap disease and may present in isolation with no or minimal muscle involvement. In East Asian populations, rapidly progressive ILD in association with clinically amyopathic DM and anti-MDA5 autoantibodies has been well described [56, 57]. In this sub-population, anti-MDA5 autoantibodies are associated with a high mortality in both adults and children [4, 58, 59]. Interestingly, a falling titre of anti-MDA5 been shown to reflect treatment response, suggesting a potential future use of some MSA in disease monitoring [60–62].
Screening for ILD with should be performed in all patients with myositis, with a minimum of a chest x-ray and pulmonary function testing. It should be remembered that x-ray is an insensitive tool in excluding ILD, and approximately 10 % of patients with ILD detected on HRCT will have a normal chest x-ray, particularly early in the disease course [63]. Clinicians should therefore have a low threshold for repeat testing and HRCT, particularly in those patients with an autoantibody profile that suggests they are at increased risk.
Skin Disease
Skin disease in DM can be a major cause of morbidity, which is particularly so in juvenile-onset disease where skin ulceration and calcinosis are more common. Although rare in adults with IIM, calcinosis is a major cause of morbidity in patients with juvenile-onset disease and occurs in up to 30 % of cases. It typically affects pressure areas such as the elbows, knees, buttocks and digits, and may lead to skin ulceration, pain from nerve entrapment and joint contractures [50, 64, 65]. In patients with juvenile-onset disease, the presence of anti-NXP2 autoantibodies substantially increases the risk of calcinosis [55]. Skin disease is also associated with the other major autoantibodies identified in juvenile-onset disease: anti-TIF1γ which is associated with skin ulceration, lipoatrophy and contractures [54, 66], and anti-MDA5 which is associated with both skin and oral ulceration [31]. In adult patients, anti-MDA5 positivity is associated with a distinct mucocutaneous phenotype characterised by skin ulceration, palmar papules, and oral pain/ulceration [26]. In the future, these patients may be managed differently, although at present there is a lack of evidence to recommend a differential treatment approach.
Treatment Approach
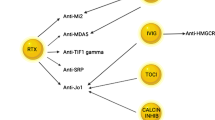
The Cochrane review of evidence-based treatment for IIM was updated in 2012, and again highlighted the lack of quality trials assessing treatment efficacy and toxicity in inflammatory myositis [67]. The heterogenicity of inflammatory myositis represents a significant barrier to the development of a general standardised treatment approach, and the autoantibody profile represents a potential method to sub-divide patients into more homogenous groups who may benefit from differing treatment approaches. There is already evidence of a differential response to B-cell depletion depending on autoantibody status in a subanalysis of the Rituximab in Myositis study, which predicted a shorter time to improvement in the presence of an antisynthetase, anti-Mi-2 or other autoantibody compared to the absence of autoantibodies [68•]. A small retrospective review also suggested a differential response in terms of relapse rate and the need for further Rituximab cycles [69].
Conclusions
The clinical utility of a test can be considered as a measure of the health care value provided by the test. Autoantibody testing in inflammatory myositis will form part of new, up-to-date diagnostic criteria. They have the potential to sub-divide patients in such a way as to highlight those at risk of significant complications and thereby guide appropriate further screening and monitoring. While the evidence base for treatment in myositis is as yet limited, it is likely that in the future autoantibody profile may also influence treatment choice.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Ceribelli A, Fredi M, Taraborelli M, Cavazzana I, Franceschini F, Quinzanini M, et al. Anti-MJ/NXP-2 autoantibody specificity in a cohort of adult Italian patients with polymyositis/dermatomyositis. Arthritis Res Ther. 2012;14(2):R97.
Bendewald MJ, Wetter DA, Li X, Davis MDP. Incidence of Dermatomyositis and Clinically Amyopathic Dermatomyositis. Arch Dermatol. 2010;146(1):26–30.
Sato S, Kuwana M. Clinically amyopathic dermatomyositis. Curr Opin Rheumatol. 2010;22(6):639–43.
Koga T, Fujikawa K, Horai Y, Okada A, Kawashiri S, Iwamoto N, et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology. 2012;51(7):1278–84.
Gono T, Kawaguchi Y, Satoh T, Kuwana M, Katsumata Y, Takagi K, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology. 2010;49(9):1713–9.
Hervier B, Devilliers H, Stanciu R, Meyer A, Uzunhan Y, Masseau A, et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev. 2012;12(2):210–7.
Phillips BA, Zilko PJ, Mastaglia FL. Prevalence of sporadic inclusion body myositis in Western Australia. Muscle Nerve. 2000;23(6):970–2.
Needham M, James I, Corbett A, Day T, Christiansen F, Phillips B, et al. Sporadic inclusion body myositis: phenotypic variability and influence of HLA-DR3 in a cohort of 57 Australian cases. J Neurol Neurosurg Psychiatry. 2008;79(9):1056–60.
Pilkington C, Tjarnlund A, Bottai M, Rider LG, Werth VP, de Visser M, et al. A47: progress report on the development of new classification criteria for adult and juvenile idiopathic inflammatory myopathies. Arthr Rheumatol. 2014;66 Suppl 11:S70–1.
Kang EH, Kuwana M, Okazaki Y, Lee EY, Lee YJ, Lee EB, et al. Comparison of radioimmunoprecipitation versus antigen-specific assays for identification of myositis-specific autoantibodies in dermatomyositis patients. Mod Rheumatol. 2014.
Musset L, Miyara M, Benveniste O, Charuel J-L, Shikhman A, Boyer O, et al. Analysis of Autoantibodies to 3-Hydroxy-3-methylglutaryl-coenzyme A Reductase Using Different Technologies. J Immunol Res. 2014. doi:10.1155/2014/405956.
Drouot L, Allenbach Y, Jouen F, Charuel J-L, Martinet JRM, Meyer A, et al. Exploring necrotizing autoimmune myopathies with a novel immunoassay for anti-3-hydroxy-3- methyl-glutaryl-CoA reductase autoantibodies. Arthritis Res Ther. 2014;16(1):1–11.
Nakashima R, Imura Y, Hosono Y, Seto M, Murakami A, Watanabe K, et al. The Multicenter Study of a New Assay for Simultaneous Detection of Multiple Anti-Aminoacyl-tRNA Synthetases in Myositis and Interstitial Pneumonia. Kuwana M, editor. PLoS ONE. 2014;9(1):e85062.
Labrador-Horrillo M, Martinez MA, Selva-O'Callaghan A, Trallero-Araguas E, Balada E, Vilardell-Tarres M, et al. Anti-TIF1 antibodies (anti-p155) in adult patients with dermatomyositis: comparison of different diagnostic assays. Ann Rheum Dis. 2012;71(6):993–6.
Casciola-Rosen L, Mammen AL. Myositis autoantibodies. Curr Opin Rheumatol. 2012;24(6):602–8. A comprehensive review of autoantibodies in myositis and their associated phenotype including autoimmune necrotising myositis.
Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology. 2009;48(6):607–12.
Hamaguchi Y, Fujimoto M, Matsushita T, Kaji K, Komura K, Hasegawa M, et al. Common and Distinct Clinical Features in Adult Patients with Anti-Aminoacyl-tRNA Synthetase Antibodies: Heterogeneity within the Syndrome. Miller F, editor. PLoS ONE. 2013;8(4):e60442.
Watanabe K, Handa T, Tanizawa K, Hosono Y, Taguchi Y, Noma S, et al. Detection of antisynthetase syndrome in patients with idiopathic interstitial pneumonias. Respir Med. 2011;105(8):1238–47.
Hervier B, Devilliers H, Stanciu R, Meyer A, Uzunhan Y, Masseau A, et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev. 2012;12(2):210–7. An analysis of phenotypic differences and prognosis between different antisynthetase autoantibodies.
Aggarwal R, Cassidy E, Fertig N, Koontz DC, Lucas M, Ascherman DP, et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis. 2013. A comparison of survival of a cohort of patients with and without anti-synthetase autoantibodies.
Huber AM, Mamyrova G, Lachenbruch PA, Lee JA, Katz JD, Targoff IN, et al. Early illness features associated with mortality in the juvenile idiopathic inflammatory myopathies. Arthritis Care Res. 2014;66(5):732–40.
Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Identification of a novel autoantibody directed against small ubiquitin-like modifier activating enzyme in dermatomyositis. Arthritis Rheum. 2007;56(9):3132–7.
Muro Y, Sugiura K, Akiyama M. Low prevalence of anti-small ubiquitin-like modifier activating enzyme antibodies in dermatomyositis patients. Autoimmunity. 2013;46(4):279–84.
Tarricone E, Ghirardello A, Rampudda M, Bassi N, Punzi L, Doria A. Anti-SAE antibodies in autoimmune myositis: identification by unlabelled protein immunoprecipitation in an Italian patient cohort. J Immunol Methods. 2012;384(1–2):128–34.
Betteridge ZE, Gunawardena H, Chinoy H, North J, Ollier WER, Cooper RG, et al. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis. 2009;68(10):1621–5.
Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): A retrospective study. J Am Acad Dermatol. 2011;65(1):25–34.
Sato S, Hoshino K, Satoh T, Fujita T, Kawakami Y, Fujita T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60(7):2193–200.
Hall JC, Casciola-Rosen L, Samedy L-A, Werner J, Owoyemi K, Danoff SK, et al. Anti-MDA5-associated dermatomyositis: Expanding the clinical spectrum. Arthritis Care Res. 2013;65(8):1307–15.
Bendewald MJ. Incidence of Dermatomyositis and Clinically Amyopathic DermatomyositisA Population-Based Study in Olmsted County. Minnesota. Arch Dermatol. 2010;146(1):26.
Gerami P, Walling HW, Lewis J, Doughty L, Sontheimer RD. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;157(4):637–44.
Tansley SL, Betteridge ZE, Gunawardena H, Jacques TS, Owens CM, Pilkington C, et al. Anti-MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: a prospective cohort study. Arthritis Res Ther. 2014;16(4):R138.
Kobayashi I, Okura Y, Yamada H. Anti-Melanoma Differentiation-Associated Gene 5 Antibody is a Diagnostic and Predictive Marker for Interstitial Lung Diseases Associated with Juvenile Dermatomyositis. J Pediatr. 2011;158(4):675–7.
Liang C, Needham M. Necrotizing autoimmune myopathy. Curr Opin Rheumatol. 2011;23(6):612–9.
Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63(3):713–21.
Amato AA, Gronseth GS, Jackson CE, Wolfe GI, Katz JS, Bryan WW, et al. Inclusion body myositis: clinical and pathological boundaries. Ann Neurol. 1996;40(4):581–6.
Brady S, Squier W, Sewry C, Hanna M, Hilton-Jones D, Holton JL. A retrospective cohort study identifying the principal pathological features useful in the diagnosis of inclusion body myositis. BMJ Open. 2014;1–14.
Salajegheh M, Lam T, Greenberg SA. Autoantibodies against a 43 KDa Muscle Protein in Inclusion Body Myositis. Chiorini JA, editor. PLoS ONE. 2011;6(5):e20266.
Pluk H, van Engelen BG, Pruijn GJM. Anti-Mup44: the first inclusion body myositis-specific autoantibody. Autoantigens, Autoantibodies. Autoimmunity. 2011;7:210.
Pluk H, van Hoeve BJA, van Dooren SHJ, Stammen-Vogelzangs J, van der Heijden A, Schelhaas HJ, et al. Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann Neurol. 2013;73(3):397–407.
Benjamin Larman H, Salajegheh M, Nazareno R, Lam T, Sauld J, Steen H, et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol. 2013;73(3):408–18.
Greenberg SA. Cytoplasmic 5′-Nucleotidase Autoantibodies in Inclusion Body Myositis: Isotypes and Diagnostic Utility. Muscle Nerve. 2014. doi:10.1002/mus.24199. An exploration of the utility of different tests for cytoplasmic 5′-nucleotidase 1A autoantibody isotypes suggests a high diagnostic sensitivity.
Aguila LA, Lopes MRU, Pretti FZ, Sampaio-Barros PD, Carlos de Souza FH, Borba EF, et al. Clinical and laboratory features of overlap syndromes of idiopathic inflammatory myopathies associated with systemic lupus erythematosus, systemic sclerosis, or rheumatoid arthritis. Clin Rheumatol. 2014;33(8):1093–8.
Koschik RW, Fertig N, Lucas MR, Domsic RT, Medsger TAJ. Anti-PM-Scl antibody in patients with systemic sclerosis. Clin Exp Rheumatol. 2012;30(2 Suppl 71):S12–6.
Labirua-Iturburu A, Selva-O'Callaghan A, Vincze M, Danko K, Vencovsky J, Fisher B, et al. Anti-PL-7 (anti-threonyl-tRNA synthetase) antisynthetase syndrome: clinical manifestations in a series of patients from a European multicenter study (EUMYONET) and review of the literature. Medicine. 2012;91(4):206–11.
Rutjes SA, Vree Egberts WTM, Jongen P, Van Den Hoogen F, Pruijn GJM, van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol. 1997;109:32–40.
Brouwer R, Hengstman GJD, Vree Egberts WTM, Ehrfeld H, Bozic B, Ghiradello A, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis. 2001;60:116–23.
Váncsa A, Csípő I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int. 2009;29(9):989–94.
La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity. 2006;39(3):249–53.
Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard JF, et al. Short-Term and Long-Term Outcome of Anti-Jo1-Positive Patients with Anti-Ro52 Antibody. Semin Arthritis Rheum. 2012;41(6):890–9.
McCann LJ, Juggins AD, Maillard SM, Wedderburn LR, Davidson JE, Murray KJ, et al. The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)–clinical characteristics of children recruited within the first 5 yr. Rheumatology. 2006;45(10):1255–60.
Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96–100.
Trallero-Araguás E, Rodrigo-Pendás JÁ, Selva-O'Callaghan A, Martínez-Gómez X, Bosch X, Labrador-Horrillo M, et al. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis: A systematic review and meta-analysis. Arthritis Rheum. 2012;64(2):523–32.
Ichimura Y, Matsushita T, Hamaguchi Y, Kaji K, Hasegawa M, Tanino Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis. 2013;71(5):710–3.
Gunawardena H, Wedderburn LR, North J, Betteridge Z, Dunphy J, Chinoy H, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology. 2007;47(3):324–8.
Tansley SL, Betteridge ZE, Shaddick G, Gunawardena H, Arnold K, Wedderburn LR, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology. 2014.
Nakashima R, Imura Y, Kobayashi S, Yukawa N, Yoshifuji H, Nojima T, et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology. 2010;49(3):433–40.
Sato S, Hirakata M, Kuwana M, Suwa A, Inada S, Mimori T, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005;52(5):1571–6.
Hoshino K, Muro Y, Sugiura K, Tomita Y, Nakashima R, Mimori T. Anti-MDA5 and anti-TIF1- antibodies have clinical significance for patients with dermatomyositis. Rheumatology. 2010;49(9):1726–33.
Kobayashi I. Interstitial lung disease associated with juvenile dermatomyositis: clinical features and efficacy of cyclosporin A. Rheumatology. 2002;42(2):371–4.
Muro Y, Sugiura K, Hoshino K, Akiyama M. Disappearance of anti-MDA-5 autoantibodies in clinically amyopathic DM/interstitial lung disease during disease remission. Rheumatology. 2012;51(5):800–4.
Gono T, Sato S, Kawaguchi Y, Kuwana M, Hanaoka M, Katsumata Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. 2012;51(9):1563–70.
Sato S, Kobayashi N, Yamazaki K, Suzuki Y. Clinical Utility of Anti-CADM-140/Melanoma differentiation-associated gene 5 autoantibody titres in patients with juvenile dermatomyositis and rapidly progressive interstitial lung disease [abstract]. Arthritis Rheum. 2012;64(10):S128.
Ryu JH, Daniels CE, Hartman TE, Yi ES. Diagnosis of Interstitial Lung Diseases. Mayo Clin Proc. 2007;62(8):976–86.
Huber AM, Feldman BM. Medium and long term functional outcomes in a mulitcenter cohort of children with juvenile dermatomyositis. Arthritis Rheum. 2000;43(3):541–9.
Rider LG, Lachenbruch PA, Monroe JB, Ravelli A, Cabalar I, Feldman BM, et al. Damage extent and predictors in adult and juvenile dermatomyositis and polymyositis as determined with the myositis damage index. Arthritis Rheum. 2009;60(11):3425–35.
Bingham A, Mamyrova G, Rother KI, Oral E, Cochran E, Premkumar A, et al. Predictors of Acquired Lipodystrophy in Juvenile-Onset Dermatomyositis and a Gradient of Severity. Medicine. 2008;87(2):70–86.
Gordon PA, Winer JB, Hoogendijk JE, Choy EHS. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database of Systematic Reviews 2012, Issue 8. Art. No.: CD003643. doi:10.1002/14651858.CD003643.pub4.
Aggarwal R, Bandos A, Reed AM, Ascherman DP, Barohn RJ, Feldman BM, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol. 2014;66(3):740–9. An analysis of patients participating in the RIM study which indicates a differential response to Rituximab dependant on autoantibody status.
Unger L, Kampf S, Luthke K, Aringer M. Rituximab therapy in patients with refractory dermatomyositis or polymyositis: differential effects in a real-life population. Rheumatology. 2014;53(9):1630–8.
Compliance with Ethics Guidelines
Conflict of Interest
Sarah L. Tansley and Neil J. McHugh declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Inflammatory Muscle Disease
Rights and permissions
About this article

Cite this article
Tansley, S.L., McHugh, N.J. Myositis Specific and Associated Autoantibodies in the Diagnosis and Management of Juvenile and Adult Idiopathic Inflammatory Myopathies. Curr Rheumatol Rep 16, 464 (2014). https://doi.org/10.1007/s11926-014-0464-1
Published:
DOI: https://doi.org/10.1007/s11926-014-0464-1