
Abstract
Subtyping myositis is helpful in the management of patients with idiopathic inflammatory myopathy (IIM). Myositis-specific and myositis-associated autoantibodies are powerful prognostic predictors of clinical manifestations, organ involvement, and outcome in this heterogeneous group of autoimmune diseases. Autoantibody assessment is helpful in managing these challenging IIM patients, including the identification of concomitant malignancy, interstitial lung disease (ILD), and refractory disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
FormalPara Key Points to Remember-
Anti-TIF1-γ, anti-NXP2, and the absence of MSA/MAA are predictive of concomitant malignancy in adults.
-
Anti-synthetase and anti-MDA5 antibodies are associated with a high risk for ILD. Anti-MDA5-associated ILD tends to be treatment-refractory requiring intensive immunosuppression, while anti-ARS-associated ILD may be more responsive to immunosuppression including glucocorticoids and rituximab.
-
Immune-mediated necrotizing myopathy, particularly in anti-SRP-positive patients, is associated with treatment-refractory disease and warrants aggressive therapy, while anti-Jo-1 and anti-Mi-2 antibodies are associated with a favorable response to rituximab.
Introduction
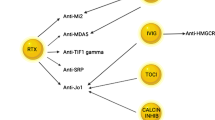
The muscular and extramuscular manifestations of the various subsets of idiopathic inflammatory myopathy (IIM) include cutaneous, gastrointestinal, pulmonary, cardiac, musculoskeletal, and vascular features. Therefore, disease subsetting or phenotyping is of vital importance for the appropriate management of myositis, particularly interstitial lung disease (ILD) and malignancy, as these complications are the leading causes of death in myositis patients [1]. Myositis-specific autoantibodies (MSAs) and myositis-associated autoantibodies (MAAs) are powerful predictive and prognostic tools regarding future clinical manifestations, treatment response, and prognosis when assessing patients and developing a management plan. Figure 19.1 incorporates autoantibody assessment in a proposed algorithm for the diagnosis and treatment of polymyositis (PM) and dermatomyositis (DM) patients.

A proposed algorithm for the management of PM/DM patients considering the MSA profile. MSAs myositis-specific autoantibodies, CADM clinically amyopathic DM, PET positron emission tomography, CT computed tomography, ILD interstitial lung disease, GGA ground-glass attenuation
Malignancy Survey
There is a well-known association of cancer with DM (and to a lesser extent PM) in up to 20% of cases [2, 3] with the diagnosis frequently being made within 1 year before or after the diagnosis of myositis [4]. Therefore, malignancy screening is essential at the time of a myositis diagnosis and the contribution of autoantibody testing is critical in stratifying the malignancy risk. In this regard, the presence of anti-TIF1-γ autoantibody or the absence of other MSAs/MAAs, including anti-Jo-1, anti-PM-Scl, anti-U1RNP, anti-U3RNP, and anti-Ku, at the time of myositis diagnosis indicates a high risk of cancer-associated myositis (CAM) [5]. In fact, this combination had a 94% sensitivity and 99% negative predictive value for the diagnosis of CAM, although the specificity and positive predictive value were only 45% and 9%, respectively for a CAM diagnosis. A meta-analysis of 312 patients with DM revealed that the sensitivity and specificity of anti-TIF1-γ antibody for the diagnosis of concomitant cancer were 78% and 89%, respectively [6], while other MSAs, most importantly anti-NXP2, and perhaps anti-SAE and anti-HMGCR positivity were also associated (to a much lesser degree) with malignancy in adult PM and DM patients [3, 5, 7,8,9,10] (Fig. 19.2). Even with anti-TIF1-γ positivity, patients over age 45 are most at risk for malignancy, as juvenile and younger adult DM patients may not be at risk for cancer. Nevertheless, clinicians should conduct an extensive malignancy survey with a diagnosis of DM and anti-TIF1-γ antibody or anti-NXP2 positivity, but the degree of cancer screening in DM patients negative for these autoantibodies remains a matter of debate. An extensive malignancy screen should include age-appropriate cancer screening; comprehensive blood tests including cancer markers; CT scans of the chest, abdomen, and pelvis; and, perhaps, a whole-body PET-CT scan in selected cases. Screening for malignancies in low-risk patients (without these high-risk antibodies) should be guided by clinical suspicion and the prevalence of individual cancers in specific ethnic groups or the country of origin. Age-appropriate screening and noninvasive tests (e.g., fecal occult blood, gynecological evaluation, prostatic-specific antigen) should be considered in all patients.

High risk for malignancy: anti-TIF1-γ, anti-NXP2, absence of an MSA/MAA.
High-risk patients should undergo extensive cancer screening and close follow-up.
Risk Stratification and Treatment of Interstitial Lung Disease
The frequency of ILD in patients with IIM is highly variable, ranging from 10% to 90%, depending on the autoantibody spectrum [11]. Moreover, the clinical course and treatment response of ILD are variable as some patients may have mild ILD responsive to glucocorticoids alone, while others may have rapidly progressive ILD (RP-ILD) resistant to intensive immunosuppressive regimens, leading to death due to respiratory failure. Clinical diagnoses are somewhat useful in predicting 5-year overall survival rates in patients with ILD: 82% in patients with PM, 71% in those with classic DM, and 59% in those with CADM [12]. However, MSAs provide much more useful information regarding frequency, severity, and treatment response in ILD. Figure 19.3 summarizes the frequency of ILD in patients with individual MSAs [8, 10, 13,14,15]. Anti-synthetase autoantibodies and anti-MDA5 are strongly linked to the presence of ILD, with a frequency approaching 90% and 50%, respectively, in the western literature. Anti-MDA5 is associated with an even higher risk in Asian countries. Although Caucasian patients with anti-SAE have a lower risk of ILD than Asian populations, the number of patients examined are too small to be confident of this association [10, 16]. The 5-year overall survival rates in patients with anti-synthetase antibodies were much better than those with anti-MDA5 (96% versus 67%) [12], as anti-MDA5 is strongly associated with RP-ILD, as shown in Fig. 19.4 [17,18,19]. Even though anti-MDA5 is clearly associated with CADM, its presence has a worse prognosis due to RP-ILD rather than the CADM clinical subset itself [20, 21]. Other risk factors for poor ILD outcomes in patients with PM/DM include skin ulcers, rapidly progressive deterioration of pulmonary function, hypoxia at diagnosis, hyperferritinemia, and ground-glass attenuation/consolidation in the lower lobe of the lung by high-resolution computed tomography (HRCT) [12, 22, 23]. Patients with an MSA associated with a high risk of ILD should undergo high-resolution CT scanning of the chest even in the absence of overt pulmonary symptoms and should be monitored for future development of ILD. In patients with an established diagnosis of ILD, one should initiate early and aggressive immunosuppressive treatment in the setting of anti-MDA5 positivity or other risk factors for ILD progression.


Anti-synthetase and anti-MDA5 autoantibodies have the highest risk of ILD.
Anti-MDA5-associated ILD is most often rapidly progressive.
Although most studies demonstrate the need for intensive immunosuppressive treatment, there is no clear evidence that one particular regimen is superior to another. High-dose glucocorticoids, in combination with immunosuppressive agents including calcineurin inhibitors, intravenous cyclophosphamide pulse therapy, and rituximab, are used in anti-MDA5-positive patients with a high risk for developing RP-ILD [24, 25]. For such patients, it is important to initiate intensive immunosuppressive regimens as early as possible. On the other hand, the short-term response to treatment with high-dose glucocorticoids is often favorable in patients with anti-synthetase autoantibodies, although ILD recurrence frequently occurs during steroid tapering necessitating additional immunosuppressive agents. In synthetase-positive patients, the choice of an immunosuppressive agent depends on severity, but rituximab is emerging as a frequently used agent [26, 27].
Anti-MDA5-associated ILD tends to be treatment refractory requiring intensive immunosuppression.
Anti-synthetase-associated ILD may be more responsive to immunosuppression including glucocorticoids and rituximab.
Refractory Myopathy
The severity of skeletal muscle involvement is quite variable, ranging from no apparent clinical myopathy (i.e., CADM) to severe disability. In some studies, DM patients with anti-TIF1-γ, anti-NXP2, and anti-SAE antibodies have more extensive myopathy (including dysphagia and severe muscle weakness) compared with subjects with an anti-synthetase, anti-MDA5, or anti-Mi-2 antibody [28,29,30]. Anti-SRP and anti-HMGCR are associated with immune-mediated necrotizing myopathy, which is often resistant to conventional immunosuppressive treatment [31]. In contrast, anti-synthetase, anti-U1RNP, anti-PM/Scl, or anti-Ku antibodies predict favorable responses to the treatment of myositis . Patients positive for MSAs linked to treatment resistance should receive glucocorticoids combined with any one of several immunosuppressive drugs, such as azathioprine, methotrexate, intravenous immunoglobulin, and rituximab (Fig. 19.1) [32,33,34,35]. Reports show that the therapeutic response to rituximab is more favorable in patients who are autoantibody positive, particularly those with anti-Jo-1 or anti-Mi-2, than in those with no MAA [26].
Immune-mediated necrotizing myopathy (particularly anti-SRP) is associated with treatment-refractory disease and should be managed aggressively, while anti-Jo-1 and anti-Mi-2 autoantibodies are associated with a favorable response to rituximab.
References
Marie I. Morbidity and mortality in adult polymyositis and dermatomyositis. Curr Rheumatol Rep. 2012;14(3):275–85.
Zampieri S, Valente M, Adami N, et al. Polymyositis, dermatomyositis and malignancy: a further intriguing link. Autoimmun Rev. 2010;9(6):449–53.
Hida A, Yamashita T, Hosono Y, et al. Anti-TIF1-γ antibody and cancer-associated myositis: a clinicohistopathologic study. Neurology. 2016;87(3):299–308.
Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96–100.
Chinoy H, Fertig N, Oddis CV, Ollier WE, Cooper RG. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis. 2007;66(10):1345–9.
Trallero-Araguás E, Rodrigo-Pendás J, Selva-O’Callaghan A, et al. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis: a systematic review and meta-analysis. Arthritis Rheum. 2012;64(2):523–32.
Allenbach Y, Keraen J, Bouvier AM, et al. High risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibody. Brain. 2016;139.(Pt 8:2131–5.
Ichimura Y, Matsushita T, Hamaguchi Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis. 2012;71(5):710–3.
Fiorentino DF, Chung LS, Christopher-Stine L, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ. Arthritis Rheum. 2013;65(11):2954–62.
Muro Y, Sugiura K, Nara M, Sakamoto I, Suzuki N, Akiyama M. High incidence of cancer in anti-small ubiquitin-like modifier activating enzyme antibody-positive dermatomyositis. Rheumatology (Oxford). 2015;54(9):1745–7.
Gono T, Kuwana M. Inflammatory myopathies: choosing the right biomarkers to predict ILD in myositis. Nat Rev Rheumatol. 2016;12(9):504–6.
Fujisawa T, Hozumi H, Kono M, et al. Prognostic factors for myositis-associated interstitial lung disease. PLoS One. 2014;9(6):e98824.
Nakashima R, Imura Y, Hosono Y, et al. The multicenter study of a new assay for simultaneous detection of multiple anti-aminoacyl-tRNA synthetases in myositis and interstitial pneumonia. PLoS One. 2014;9(1):e85062.
Hamaguchi Y, Kuwana M, Hoshino K, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011;147(4):391–8.
Suzuki S, Nishikawa A, Kuwana M, et al. Inflammatory myopathy with anti-signal recognition particle antibodies: case series of 100 patients. Orphanet J Rare Dis. 2015;10:61.
Ge Y, Lu X, Shu X, et al. Clinical characteristics of anti-SAE antibodies in Chinese patients with dermatomyositis in comparison with different patient cohorts. Sci Rep. 2017;7(1):188.
Sato S, Murakami A, Kuwajima A, et al. Clinical utility of an enzyme-linked immunosorbent assay for detecting anti-melanoma differentiation-associated gene 5 autoantibodies. PLoS One. 2016;11(4):e0154285.
Chen Z, Cao M, Plana MN, et al. Utility of anti-melanoma differentiation-associated gene 5 antibody measurement in identifying patients with dermatomyositis and a high risk for developing rapidly progressive interstitial lung disease: a review of the literature and a meta-analysis. Arthritis Care Res (Hoboken). 2013;65(8):1316–24.
Hozumi H, Fujisawa T, Nakashima R, et al. Comprehensive assessment of myositis-specific autoantibodies in polymyositis/dermatomyositis-associated interstitial lung disease. Respir Med. 2016;121:91–9.
Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60(7):2193–200.
Moghadam-Kia S, Oddis CV, Sato S, et al. Anti-melanoma differentiation-associated gene 5 is associated with rapidly progressive lung disease and poor survival in US patients with amyopathic and myopathic dermatomyositis. Arthritis Care Res (Hoboken). 2016;68(5):689–94.
Gono T, Kawaguchi Y, Hara M, et al. Increased ferritin predicts development and severity of acute interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49(7):1354–60.
Tanizawa K, Handa T, Nakashima R, et al. The prognostic value of HRCT in myositis-associated interstitial lung disease. Respir Med. 2013;107(5):745–52.
Nakashima R, Hosono Y, Mimori T. Clinical significance and new detection system of autoantibodies in myositis with interstitial lung disease. Lupus. 2016;25(8):925–33.
So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5antibody-positive amyopathic dermatomyositis. Clin Rhumatol. 2018;37(7):1983–9.
Aggarwal R, Bandos A, Reed AM, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol. 2014;66(3):740–9.
Allenbach Y, Guiguet M, Rigolet A, et al. Efficacy of rituximab in refractory inflammatory myopathies associated with anti- synthetase auto-antibodies: an open-label, Phase II trial. PLoS One. 2015;10(11):e0133702.
Mugii N, Hasegawa M, Matsushita T, et al. Oropharyngeal dysphagia in dermatomyositis: associations with clinical and laboratory features including autoantibodies. PLoS One. 2016;11(5):e0154746.
Rogers A, Chung L, Li S, Casciola-Rosen L, Fiorentino DF. The cutaneous and systemic findings associated with nuclear matrix protein-2 antibodies in adult dermatomyositis patients. Arthritis Care Res (Hoboken). 2017;69:1909.
Fujimoto M, Watanabe R, Ishitsuka Y, Okiyama N. Recent advances in dermatomyositis-specific autoantibodies. Curr Opin Rheumatol. 2016;28(6):636–44.
Watanabe Y, Uruha A, Suzuki S, et al. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. J Neurol Neurosurg Psychiatry. 2016;87(10):1038–44.
Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65(2):314–24.
Valiyil R, Casciola-Rosen L, Hong G, Mammen A, Christopher-Stine L. Rituximab therapy for myopathy associated with anti-signal recognition particle antibodies: a case series. Arthritis Care Res (Hoboken). 2010;62(9):1328–34.
Ramanathan S, Langguth D, Hardy TA, et al. Clinical course and treatment of anti-HMGCR antibody-associated necrotizing autoimmune myopathy. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e96.
Marie I, Menard JF, Hatron PY, et al. Intravenous immunoglobulins for steroid-refractory esophageal involvement related to polymyositis and dermatomyositis: a series of 73 patients. Arthritis Care Res (Hoboken). 2010;62(12):1748–55.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gono, T., Kuwana, M. (2020). Role of Myositis Autoantibodies in Management and Prognosis. In: Aggarwal, R., Oddis, C. (eds) Managing Myositis. Springer, Cham. https://doi.org/10.1007/978-3-030-15820-0_19
Download citation
DOI: https://doi.org/10.1007/978-3-030-15820-0_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-15819-4
Online ISBN: 978-3-030-15820-0
eBook Packages: MedicineMedicine (R0)