
Abstract
Rheumatoid arthritis (RA) is associated with increased cardiovascular (CV) morbidity and mortality, related not only to traditional CV risk factors, but also to a chronic inflammatory state. However, lipid profiles in RA are different from those observed in the general population at risk of CV disease, where there is evidence of a positive relationship between disease and high cholesterol levels. In untreated patients with active RA this relationship is different, with a paradoxical effect resulting in lower levels of cholesterol associated with an increased risk of CV disease. In this review, we summarize the latest evidence on lipid abnormalities in the setting of RA and the interaction between inflammation and lipoproteins, as well as the effect of DMARDs and biologic therapies on lipid profiles and the possible implications for CV outcomes in this population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In inflammatory rheumatic diseases, accelerated atherosclerosis leads to increased cardiovascular (CV) morbidity and mortality [1]. Patients with rheumatoid arthritis (RA) have higher odds ratios (ORs) for ischemic cerebro-vascular events and myocardial infarction (MI), as compared with healthy matched controls [2, 3]. This increased CV risk remains after adjusting for traditional cardiac risks factors, including age, diabetes, hyperlipidemia, and hypertension [4].
The lipid profile in patients with RA differs from what is observed in the population at large. Many studies have reported decreases in total cholesterol (TC), low-density lipoprotein cholesterol (LDLc), and high-density lipoprotein cholesterol (HDLc) prior to RA development and in patients with active untreated RA [5–7]. Since both HDLc and TC are decreased, the atherogenic index (AI), calculated as the TC/HDLc ratio, will vary according to their relative decrease; in fact, high AI has been reported despite low TC [7]. These findings contrast with those in patients without RA, where CV risk is associated with increased cholesterol levels [8]. In RA, the major determinant of CV disease appears to be the presence of elevated inflammatory markers [9•].
These paradoxical effects are not well understood, and the relationship and interactions between inflammation, lipid profiles, and CV disease in RA appear to be quite complex. Furthermore, the effects of RA therapies on lipid profiles and CV risk as inflammation decreases are only recently being recognized. We did a literature search in Medline, EMBASE, and the Cochrane Library of articles in English, French, German, and Spanish published between January 2007 and March 2012 on the most recent evidence on lipid profiles in RA, the interaction between inflammation and lipoproteins, the effects of disease-modifying antirheumatic drugs (DMARD) and biologic therapies on lipid profiles, and the impact of these factors on CV disease.
Atherosclerosis
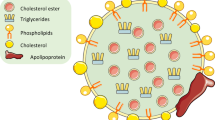
In individuals without RA, one of the major contributors to atherosclerosis and CV events is an atherogenic lipid profile with high levels of TC and LDLc, together with low levels of HDLc [8]. Various mechanisms are involved in the pathogenesis of the atherosclerotic lesion, including inflammation, dyslipidemia, endothelial damage, and immune processes. Figure 1 schematically shows the mechanisms involved in atherosclerosis. Briefly, LDLc moves in and out of artery walls, where it is oxidized (oxLDLc), becoming proinflammatory. Endothelial cells are injured, increasing the expression of adhesion molecules and releasing inflammatory products, particularly cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), interleukin-6 (IL-6), and interferon gamma (INF-γ). The release of cytokines attracts monocytes into the artery wall, crossing it and evolving into monocytes/macrophages that engulf oxLDLc to form foam cells. These cells play a key role in the formation of the atherosclerotic plaque by releasing growth factors and proteinases that destroy normal tissue and cause smooth muscle hypertrophy in the arterial wall. Cytokines released by damaged endothelium and foam cells also attract additional cells, including T and B lymphocytes, which contribute to further damage. HDLc protects the artery wall from plaque formation through its apolipoprotein A (ApoA-I) molecules, which help in the reverse transport of cholesterol. ApoA-I can move cholesterol from foam cells into macrophages and out of the endothelial cells. Moreover, HDLc inhibits oxidation of LDLc through the release of antioxidant enzymes such as lecithin cholesterol acyltransferase (LCAT) and paroxonase 1 (PON1). HDLc has also been implicated as an immunomodulator through inhibition of adhesion molecule expression in endothelial cells [10, 11]. HDLc molecules are altered during stress and inflammation, with decreased levels of Apo A and PON1 as a result of the oxidative stress process, losing some of its anti-inflammatory properties (pro-inHDLc) [10].

Pathogenesis of atherosclerosis (1) Cytokines attract monocytes (Mono) ,damaging endothelial cells (ECs) ,increasing adhesion molecules (AM) and inflammation. (2) and (3) Monocytes evolve into macrophages (M) incorporating LDLc through the LDLc receptor (LDLcR). (4) LDLc is oxidized (oxLDLc), becoming pro-inflammatory. Macrophages engulf oxLDLc to form foam cells (FCs). Damaged ECs increase AMs and release inflammatory products: tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), interleukin-6 (IL-6), and Interferon gamma (INF-γ). (5) FCs release growth factors and proteinases, damaging tissue and leading to atherosclerotic plaque formation. (6) Cytokines attract additional cells, including T and B cells, promoting further damage. (7) Interaction between T and B cells perpetuates inflammation. Right upper corner of figure: HDLc prevents atherosclerosis, through the role of apolipoprotein A (ApoA-I) in the reverse transport of cholesterol from FC to M, and out of EC in the arterial wall, and from interfering with the oxidation of LDLc by antioxidant enzymes, including Lecithin/cholesterol acyltransferase (LCAT) and paroxonase 1 (PON1)
Lipid Profiles in RA
The lipid profile in RA is altered quantitatively and qualitatively. A continuous decline in TC and LDLc levels has been demonstrated during the 5 years prior to the development of RA, and an increase in the AI has also been observed [5–7]. Different observational studies have found abnormal lipoprotein and apolipoprotein patterns in RA, with higher levels of lipoprotein a [Lp (a)]. Lp (a) is a cholesterol-rich lipoprotein similar to the LDLc particle, known to be a risk factor for atherosclerosis [12]. HDLc is decreased with an increase in TC/HDLc and LDLc/HDLc ratios. Apolipoprotein B (Apo B) is one of the major apolipoproteins in LDLc; the Apo B/Apo A ratio is also increased in RA [7, 13, 14]. There is controversy regarding triglyceride (TG) levels in RA, with some suggesting an increment in TG in the setting of inflammation [15] and others reporting decreased levels [16].
While studies in the general population indicate a positive relationship between CV disease and cholesterol levels [8], in RA this relationship is different, likely as a result of a chronic inflammatory state, where inflammation as a CV risk factor seems to be inversely associated with cholesterol levels [9•]. In the CARRE study, which examined the occurrence of fatal and nonfatal CV events and their association with lipid profiles and inflammatory markers in a cohort of patients with RA, increasing C-reactive protein (CRP), a well known inflammatory marker, was negatively associated with TC, HDLc, and Apo A-I levels [17]. Inflammatory markers are associated with increased risk of CV death in RA patients, even when other CV risk factors are controlled for [18]. In a population-based inception cohort of patients with RA, Myasoedova et al. found that CV risk in RA was associated with an increased erythrocyte sedimentation rate (ESR) and, concomitantly, decreased TC and LDLc [19••].
Effect of Therapy on the Lipid Profiles of Patients With RA
There are different targets in the therapeutic pathophysiologic pathways in RA. Nonsteroidal anti-inflammatory drugs (NSAIDS) are used for symptomatic control, and although relevant to CV outcomes, their effects are beyond the topic of this review. We will review here the effects of treatment with (1) glucocorticoids, (2) traditional DMARDs, and (3) biologic agents. In general, it is known that as inflammation decreases with therapy in RA, lipid levels rise [20–22], and TC and HDLc appear to increase more in DMARD responders, as compared with nonresponders [23]. A summary of the reported effects on lipids of the various RA therapies is shown in Table 1. Specific findings for each drug or class of drugs are described below.
Glucocorticoids
The current thinking about the use of glucocorticoids in RA is that they adversely impact lipid profiles and CV risk [24]. Yet there is contradictory evidence on the impact of steroids on lipid metabolism; many studies have not adequately controlled for confounding factors, and often, the dosages used in many studies is higher than the low doses recommended for therapy in RA [25]. In a randomized controlled trial evaluating the effect of low-dose prednisolone (7.5 mg daily) added to DMARD, Hafstrom et al. [26] examined the lipid profiles of patients after treatment for 2 years and did not find statistically significant differences in the levels of TC, LDLc, HDLc, Lp (a), and TG between those who received steroids and those who did not. However, in a subset of patients who were treated with prednisolone for 4 years, there was a significant increase in TC. Supporting these results, a cross-sectional study in women with RA [27] examined lipid profiles, comparing patients treated with low-dose prednisone (mean dose, 5.1 mg daily) in conjunction with DMARDs for a mean total duration of therapy of 9 years with patients treated with DMARDs only. There were no changes in the level of TC, TG, LDLc, and its main protein, Apo B, in patients on low-dose steroid therapy, but HDLc levels increased by 14.7 %. This suggests that in patients with RA, the corticosteroid effect of suppressing inflammation may be more important than the effects on lipid profiles when inflammation is not present [17]. In another study, glucocorticoid therapy was independently correlated with increased levels of HDLc and TG, but not with levels of TC and LDLc; CRP levels had an inverse relationship with TC, HDLc, and LDLc levels, but not with any lipid ratio [28•, 29]. These findings diverge from the observation made by Chung et al. [30••], where higher ORs of high LDLc were found in patients with RA taking steroids, as compared with controls (multivariable-adjusted OR = 2.12 {1.14, 3.95}).
Independently of the effect of steroids on the lipid profiles of patients with RA, these agents increase the prevalence of hyperglycemia, diabetes mellitus type 2, osteoporosis, and hypertension [31–34]. The use of steroids in high cumulative doses has been linked to increased risk of CV mortality, MI, and heart failure [35].
Because the interplay between inflammation and lipid metabolism is complex, it is reasonable to think that lipid profiles and CV risk of patients with RA treated with steroids result from the opposing effects of the anti-inflammatory action of steroids, and the potential resulting decrease in atherosclerosis, and their deleterious effects on other CV risk factors, including hypertension, diabetes mellitus type 2, and hyperlipidemia. The final CV risk and outcome will depend on which of these effects prevail, also considering duration of treatment and dose [25–27].
DMARDS
As was mentioned before, a decrease in inflammation can result in lower CV risk, independently of the effects of abnormal lipid profiles. However, individual DMARDs may have added effects. Hydroxychloroquine (HCQ) is an antimalarial drug with DMARD properties commonly used in RA and systemic lupus erythematosus [14]. Munro et al. [36] compared patients with RA treated with HCQ or gold and observed at 12 months a 15 % rise in HDLc in those treated with HCQ, as compared with a 12 % decline in those treated with gold (p = .006). TC levels remained unchanged in both groups. The mechanisms underlying the beneficial effects of HCQ on lipid profiles are not clear. While it may have a general effect through the reduction of inflammation and disease activity, it may also decrease cholesterol synthesis in the liver and up-regulate LDLc receptors. Other research has also supported the beneficial effects of HCQ in lipid profiles [29], and a recent study by Toms et al. [28•] showed that HCQ use was inversely associated with higher levels of TC and LDLc and lower TC/HDLc and LDLc/HDLc ratios in RA patients. Consistent with these results, Morris et al. [37] found, in a retrospective study, that HCQ use was associated with a significant decrease in LDLc, TC, LDLc/HDLc ratio, and TC/HDLc (AI) in patients with RA.
With respect to CV outcomes, there is no data to support whether the favorable changes on lipids seen with HCQ translate into better CV outcomes. A concern for cardiomyotoxicity has been raised on the basis of case reports (at least 20) of patients suffering from restrictive cardiomyopathy and arrhythmias after prolonged use of chloroquine or HCQ [38, 39].
Methotrexate (MTX) is the cornerstone treatment for RA. Data regarding the impact of MTX on lipid profiles are sparse and conflicting. In an observational study [29] in patients with RA taking MTX or leflunomide, there was no significant difference in the levels of LDLc, HDLc, and TG. In contrast, another study reported that MTX may induce increments in both TC and TG levels [40]. Another report showed higher levels of TC and HDLc associated with use of MTX [28•]. A recent systematic review [41] evaluated the effects of MTX on CV disease in RA patients: One study found no differences in lipid profiles after a 1-year treatment with MTX; another one reported improvement in lipid profiles, correlating with changes in CRP and ESR levels; finally, another found no changes in lipid profiles with reductions in CRP during MTX therapy. Given the conflicting results in these studies, it is difficult to establish the net effect of MTX on lipids in these patients. With regard to CV disease in patients with RA, most of the studies evaluated in this review [41] showed that CV morbidity, including death, MI, heart failure, and ischemic stroke, were significantly decreased in patients taking MTX. Overall, regardless of the conflicting effect of MTX on lipid profiles in patients with RA, there seem to be beneficial CV outcomes in this population, as supported by another study demonstrating a reduced rate of MI in association with MTX therapy in patients with RA [42].
Tumor Necrosis Factor α (TNF-α) Inhibitors
TNF-α is an inflammatory cytokine that has an atherogenic effect on the endothelial cells of artery walls, inducing cell apoptosis and up-regulating expression of adhesion molecules [43]. TNF-α inhibitors neutralize soluble and/or membrane-bound TNF-α. Currently, there are five agents approved for the treatment of RA in the US: infliximab (human–mouse chimeric antibody), etanercept (fully humanized TNF receptor-IgG1 fusion protein), adalimumab (recombinant humanized IgG1 monoclonal antibody), certolizumab-pegol (novel PEGylated human anti-TNF-α antibody), and golimumab (fully human anti-TNF-α monoclonal antibody) [44]. Our prior systematic review [45] found an increase in levels of TC, LDLc, HDLc, and TG with treatment with anti-TNF-α in patients with RA. These data are also supported by the subsequent literature, including a recent review by van Sijl et al. [46] that found that TC levels rose up to 10 % and HDLc increased up to 7 % within 6 months of therapy with TNF-α inhibitors, without changes in the AI. Another meta-analysis [47] also suggested that anti-TNF-α therapy is associated with significant increments of TC, HDLc, and TG levels; LDLc and AI were not affected. With long-term therapy, the ApoB/ApoA ratio seemed to decrease, but further evaluation of these results is necessary in larger studies.
A potential benefit from anti-TNF-α therapy is its qualitative effect on HDLc. Oxidative changes seen during inflammation and the presence of serum amyloid A alter HDLc structure and decrease Apo A-I, an effect reversed with TNF-α inhibitors [11, 48]. The beneficial effect of TNF-α inhibitors could also be related to the suppression of inflammation, with decreased CRP levels associated, in turn, with increments in HDLc [20]. The overall rise in TC levels that is seen after 6 weeks of treatment can be a consequence of an increase in HDLc, with a trend toward a more athero-protective lipid profile that can potentially represent a benefit in terms of CV risk in patients with RA [20, 45].
There is evidence to support that anti-TNF-α therapy reduces the incidence of cerebro-vascular events, heart failure (HF), and MI in patients with RA [48, 49]. However, patients with heart failure classes III and IV, on the basis of New York Heart Association (NYHA) criteria, have a contraindication for anti-TNF-α therapy because of an increased risk of worsening HF [50].
T-Cell Costimulatory Modulation
Abatacept is a recombinant fully human soluble fusion protein used in the treatment of RA that modulates costimulation of T-lymphocytes and prevents activation of B-lymphocytes and macrophages, resulting in decreased levels of proinflammatory cytokines and complement fixation [48, 51]. Data on the impact of abatacept on the lipid profile of patients with RA are scarce. Phase III trials of abatacept combined with DMARDS, and compared with DMARDS alone, found no significant differences in the incidence of MI, cardiac arrest, hypertension, and HF, as compared with patients treated with placebo and DMARDs [48].
Rituximab
Few studies have evaluated the effect of CD20 targeted therapy on lipids. Rituximab is a human-murine chimeric monoclonal antibody against the B-lymphocyte specific CD20 surface protein, approved for the treatment of refractory RA. One study examined the lipid profiles of 5 patients with RA refractory to anti-TNF-α agents treated with rituximab and followed for 16 weeks. They observed a 3 %–11 % decrement in TC and a 14 %–35 % increment in HDLc levels. Variable levels of TG were found, with a net effect of zero by week 16 [52]. In a study carried out by Gonzalez-Juanatey et al. [53] evaluating the effect of rituximab on endothelial dysfunction and lipid profiles in 6 patients with RA, no significant differences were observed in TC, LDLc, HDLc, and TG after 2 weeks and 6 months of therapy. A potential athero-protective effect of rituximab is the decrease in proatherogenic IgG antibodies against oxLDLc, as well as decreased secretion of INF-γ by depletion of B-lymphocytes that causes a modulatory effect on T-cells. Further investigations are needed to confirm these effects on a long-term basis, in larger samples of patients, and to evaluate their impact on CV outcomes in patients with RA [54].
Available research evaluating the CV impact of rituximab in patients with RA is scant, but the most common findings are related to the infusion itself with hypotension and edema and, very rarely but fatally, with MI, and with life-threatening arrhythmias [48, 55]. Van Vollenhoven did not find an increased frequency of MI in RA patients treated with rituximab when comparing them with the general population [56].
Tocilizumab
Interleukin 6 (IL-6) is an inflammatory cytokine with effects in multiple systems, including immune, hepatic, and hematologic, and is considered the major regulator of CRP synthesis in the liver. IL-6 is elevated in RA [57], and persistent elevated levels of CRP are associated with CV morbidity [58] and all cause mortality [59]. Tocilizumab, a recombinant human monoclonal antibody, blocks soluble and membrane-bound IL-6 receptors [60]. In patients with RA, this agent has an adverse impact in lipid profile [48, 61]. A recent meta-analysis [62] found elevations of TC, LDLc, and HDLc during the first weeks of treatment that remained up to several years during follow-up, without changes in the AI. A subsequent study of 9 patients with RA who received tocilzumab reported similar results, with increases in TC, LDLc, HDLc, Apo A-I, and Apo A-2 and no effect on the AI [63]. However, until now, there has been no evidence supporting an increase in CV events in patients treated with tocilizumab [48, 62, 63].
Statins and RA
Statins have recognized beneficial effects in the primary and secondary prevention of CV events in the general population with lipid abnormalities [64]. The clinical benefits of statins are attributed to multiple effects, including anti-inflammatory actions preventing endothelial dysfunction and a decrease in the oxidation of lipids, especially LDLc [65]. Patients with RA have as much benefit from therapy with statins as lipid-lowering agents as the general population, with cardio-protective effects noted. This was seen in the IDEAL trial, which examined the effect of statin therapy on lipid levels and CV mortality and morbidity in a subgroup of patients with RA and previous MI [66]. There is also limited evidence suggesting that discontinuation of statin therapy in patients with RA for more than 3 months is associated with increased risk of myocardial infarction [67]. Toms et al. [68], in a recent observational study, raised a concern for underuse of statins in patients with RA without CVD; these patients were stratified by different risk scores as at high risk of CV events, and depending on the method of risk stratification, up to 26 % of patients with RA should have been receiving statin therapy for primary CV prevention, but the use of this drug was suboptimal in this population.
El-Barbary et al. [69] evaluated disease activity, inflammatory markers, and lipid profiles in 30 patients with early RA (less than 1 year of disease duration). All of them were treated with MTX (mean dose, 15.5 mg weekly) and prednisone (10 mg/day), but only 15 of them received atorvastatin (40 mg/day); both groups were compared with 10 matched healthy controls. A statistically significant increment in HDLc was observed, with a decrease in LDLc, TG levels, and AI, in association with significant improvement in disease activity scores and reduction in inflammatory markers, including ESR and CRP, in both groups, but much more prominently in patients receiving atorvastatin. This study supports the findings of the TARA trial [70], where, in a double-blind placebo-controlled study with intention to treat, 116 patients with RA were randomized into two groups, one treated with atorvastatin (40 mg/day) and DMARDs, and the other one with DMARDs and placebo, followed for 6 months. A modest but clinically and statistically relevant effect was observed for patients receiving atorvastatin, with decreased disease activity and declines in CRP and ESR at 6 months of 50 % and 28 %, respectively.
Interestingly, a retrospective cohort study by Jick et al. [71], using the General Practice Research Database in the U.K., found that in patients with hyperlipidemia, those who were prescribed statins had lower risk of developing RA, as compared with those who did not.
Little is known with regard to the interaction between DMARDS, biologic drugs, and concomitant use of statins. Of interest, studies by Winiarska et al. [72] and Arts et al. [73] investigated whether the concomitant use of rituximab with statins inhibited the effect of this drug in RA activity. Patients on statins had a statistically significant shorter effective treatment period, as compared with patients not exposed to statins. This could be related to changes in the structure of CD20 induced by statins, which could alter the capability of rituximab to bind to this receptor [72]. More research in this area is necessary to validate these observations.
Current recommendations for CV risk management in patients with RA [74] state that statin therapy should be implemented when cholesterol levels are elevated, mostly targeting LDLc, as is done in the general population, and after estimating CV risk individually [8, 75, 76]. However, it has been suggested that risk score models for the general population should be adapted for patients with RA, increasing risk by 1.5 when the patient meets two or more of these factors: presence of certain extra-articular manifestations, positive rheumatoid factor or anti-cyclic citrullinated peptide antibody, or disease duration of 10 years or more [74].
Other Aspects of Lipid and CV Risk Management in Patients With RA
Patients with RA are at increased risk of CV morbidity and mortality. In addition to the risk conferred by a persistent inflammatory state, it has also been shown that patients with RA have increased rates of undiagnosed type 2 diabetes mellitus and hypertension [30••]. Treatment target goals are accomplished only in 57 % of RA patients with diabetes and in 40 % of RA patients with hypertension [30••].
Some studies suggest that in patients with RA treated with DMARDS and/or biologics drugs, the assessment of lipid profiles should be focused on lipid ratios rather than individual lipoproteins, given that the ratios tend to be less affected by RA treatment [17, 28•]. However, further longitudinal studies are necessary to determine the best approach in the long-term management of these patients.
Conclusions
Lipid metabolism in chronic inflammatory diseases is complex. In RA, the paradoxical relationship between lipid profiles and CV risk might be mediated by the effects of inflammation. While many agents commonly used for the treatment of RA can alter lipid profiles with an increase in TC, these changes are often accompanied by increases in HDLc and minimal impact on deleterious lipid ratios. Overall, it appears that tight therapeutic control of inflammation can decrease CV risk and related deleterious outcomes. Additional research is needed to better establish the pathophysiology of lipid abnormalities in RA, the pharmacologic effects of RA therapy on CV risk, and the potential effects of drugs such as statins that are beneficial for the CV risk of the population at large.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
John H, Toms TE, Kitas G. Rheumatoid arthritis: is it a coronary heart disease equivalent? Curr Opin Cardiol. 2011;26:327–33.
Watson DJ, Rhodes T, Gues HA. All cause mortality and vascular events among patients with rheumatoid arthritis, osteoarthritis or no arthritis in the UK General Practice Research Database. J Rheumatol. 2003;30:1196–202.
Semb AG, Kvien TK, Aastveit AH, et al. Lipids, myocardial infarction and ischemic stroke in patients with rheumatoid arthritis in the Apolipoprotein- related Mortality RISK (AMORIS) Study. Ann Rheum Dis. 2010;69:1996–2001.
Del Rincon ID, Williams K, Stern MP, et al. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional risks factors. Arthritis Rheum. 2001;44:2737–45.
Myasoedova E, Kremers HM, Fits-Gibbon P, et al. Lipid profile improves with the onset of rheumatoid arthritis. Ann Rheum Dis. 2009;68 Suppl 3.
Myasoedova E, Crowson CS, Kremers HM, et al. Total cholesterol and LDL levels decrease before rheumatoid arthritis. Ann Rheum Dis. 2010;69:1310–4.
Park YB, Lee SK, Lee WK, et al. Lipid profiles in untreated patients with rheumatoid arthritis. J Rheumatol. 1999;26:1701–4.
Anderson KM, Castelli WP, Levy D. Cholesterol and mortality: 30 years of follow-up from the Framingham study. JAMA. 1987;257:2176–80.
• Choy E, Sattar N. Interpreting lipid level in the context of high grade inflammatory states with a focus on rheumatoid arthritis: a challenge to conventional cardiovascular risk actions. Ann Rheum Dis. 2009;68:460–569. Th is article explains the relationship between inflammation and lipid profiles, why lipoproteins are altered in patients with RA, and how these changes can translate into clinical relevant CV risk factors.
Hahn BH, Grossman J, Chen WC, et al. The pathogenesis of atherosclerosis in autoimmune rheumatic diseases: roles of inflammation and dyslipidemia. J Autoimmun. 2007;28:69–75.
Hahn BH, Grosmman J, Benjamin JA, et al. Altered lipoprotein metabolism in chronic inflammatory states: proinflammatory high-density lipoprotein and accelerated atherosclerosis in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res Ther. 2008;10:213.
Dahlen GH. Lp (a) lipoprotein in cardiovascular disease. Atherosclerosis. 1994;108:111–26.
Yoo WH. Dyslipoproteinemia in patients with active rheumatoid arthritis: Effect of disease activity, sex, and menopausal status on lipid profiles. J Rheumatol. 2004;31:1746–53.
Toms T, Symmons D, Kitas G. Dyslipidaemia in rheumatoid arthritis: the role of inflammation, drugs, lifestyle and genetic factors. Curr Vasc Pharmacol. 2010;8:301–26.
Dursunoglu D, Evrengul H, Polat B, et al. Lp (a) lipoprotein and lipids in patients with rheumatoid arthritis: serum levels and relationship to inflammation. Rheumatol Int. 2005;25:241–5.
Rantapaa-Dahlqvist S, Wallberg-Jonsson S, Dahlen G. Lipoprotein (a), lipids, and lipoproteins in patients with rheumatoid arthritis. Ann Rheum Dis. 1991;50:366–8.
Peters MJL, Voskuyl AE, Sattar BA, et al. The interplay between inflammation, lipids and cardiovascular risk in rheumatoid arthritis: why ratios may be better. Int J Clin Pract. 2010;64:1440–3.
Maradit-Kremers H, Nicola PJ, Crowson CS, et al. Cardiovascular death in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2005;52:722–32.
•• Myasoedova E, Crowson CS, Kremers HM, et al. Lipid paradox in rheumatoid arthritis: the impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann Rheum Dis. 2011;70:482–7. In this study, concrete evidence of the paradoxical relationship between lower levels of TC and LDLc and increased CV risk in patients with RA is demonstrated, as well as the interplay between inflammation as a CV risk factor.
Peters MJ, Vis M, van Halm VP, et al. Changes in lipid profile during infliximab and corticosteroid treatment in rheumatoid arthritis. Ann Rheum Dis. 2007;66:958–61.
Steiner G, Urowitz MB. Lipid profiles in patients with rheumatoid arthritis: mechanisms and the impact of treatment. Semin Arthritis Rheum. 2009;38:372–81.
Schimmel EK, Yazici Y. Increased lipid levels but unchanged atherogenic index in rheumatoid arthritis patients treated with biologic disease modifying antirheumatic drugs: published experience. Clin Exp Rheumatol. 2009;27:446–51.
Park YB, Choi SJ, Kim MY, et al. Effects of anti-rheumatic therapy on serum lipid levels in patients with rheumatoid arthritis: a prospective study. Am J Med. 2002;113:188–93.
Maxwell SR, Moots RJ, Kendall MJ. Corticosteroids: do they damage the cardiovascular system? Postgrad Med. 1994;70:863–70.
Bruce IN. “Not only.. but also”: factors that contribute to accelerated atherosclerosis and premature coronary heart disease in systemic lupus erythematosus. Rheumatology (Oxford). 2005;44:1492–502.
Hafstrom I, Rohani M, Deneberg S, et al. Effects of low-dose prednisolone on endothelial function, atherosclerosis, and traditional risk factors for atherosclerosis in patients with Rheumatoid arthritis: a randomized study. J Rheumatol. 2007;34:1810–6.
Garcia-Gomez C, Nolla JM, Valverde J, et al. High HDL-cholesterol in women with rheumatoid arthritis on low-dose glucocorticoid therapy. Eur J Clin Invest. 2008;38:686–92.
• Toms T, Panoulas V, Douglas K, et al. Are lipid ratios less susceptible to change with systemic inflammation than individual lipid components in patients with rheumatoid arthritis? Angiology. 2011;62:167–75. This study provides evidence on how, in the setting of inflammation, lipid ratios are less susceptible to changes induced by most antirheumatic drugs. Using these biomarkers as a more reliable method to evaluate lipid abnormalities can assist clinicians in their estimates of CV risk in patients with RA and in the management of traditional risk factors.
Rho YH, Oeser A, Chung C, et al. Drugs used in the treatment of rheumatoid arthritis: relationship between current use and cardiovascular risk factors. Arch Drug Info. 2009;2:34–40.
•• Chung C, Petri M, Post W, et al. Prevalence of traditional modifiable cardiovascular risk factors in patients with rheumatoid arthritis: comparison with control subjects from the Multi-Ethnic Study of atherosclerosis. Semin Arthritis Rheum. 2012;41:535–44. This paper stresses the importance of controlling traditional CV risk factors in patients with RA. It shows that patients with RA have high rates of undiagnosed diabetes mellitus type 2, hypertension, and elevated LDLc and that fewer than 60 % of them are able to achieve preestablished target goals for each of these conditions.
Ravindran V, Rachapalli S, Choy EH. Safety of medium to long term glucocorticoid therapy in rheumatoid arthritis: a meta-analysis. Rheumatology (Oxford). 2009;48:807–11.
Loddenkemper K, Bohl N, Perka C, et al. Correlation of different bone markers with bone density in patients with rheumatic diseases on glucocorticoid therapy. Rheumatol Int. 2006;26:331–6.
Panoulas VF, Douglas KM, Stavropoulos-Kalinoglou A, et al. Long-term exposure to medium-dose glucocorticoid therapy associates with hypertension in patients with rheumatoid arthritis. Rheumatology (Oxford). 2008;47:72–5.
Hoes JN, Jacobs JW, Hulsmans HM, et al. High incidence rate of vertebral fractures during chronic prednisone treatment, in spite of bisphosphonate or alfacalcidol use: extension of the alendronate or alfacalcidol in glucocorticoid-induced osteoporosis-trial. Clin Exp Rheumatol. 2010;28:354–9.
Davis 3rd JM, Maradit-Kremers H, Crowson CS, et al. Glucocorticoids and cardiovascular events in rheumatoid arthritis: a population- based cohort study. Arthritis Rheum. 2007;56:820–30.
Munro R, Morrison E, McDonald AG, et al. Effect of disease modifying agents on the lipid profiles of patients with rheumatoid arthritis. Ann Rheum Dis. 1997;56:374–7.
Morris SJ, Wasko MC. Antohe Jl, et al. Hydroxychloroquine use is associated with improvement in lipid profiles in rheumatoid arthritis. Arthritis Care Res. 2011;63:530–4.
Cotroneo J, Sleik KM, Rene Rodriguez E, et al. Hydroxychloroquine-induced restrictive cardiomyopathy. Eur J Echocardiogr. 2007;8:247–51.
Naqvi TZ, Luthringer D, Marchevsky A, et al. Chloroquine induced cardiomyopathy-echocardiographic features. J Am Soc Echocardiogr. 2005;18:383–7.
Saiki O, Takao R, Naruse Y, et al. Infliximab but not methotrexate induces extra-high levels in VLDL triglyceride in patients with rheumatoid arthritis. J Rheumatol. 2007;34:1997–2004.
Westlake S, Colebatch A, Baird J, et al. The effects of methotrexate on cardiovascular disease in patients with rheumatoid arthritis: a systematic literature review. Rheumatology. 2010;48:295–307.
Suissa S, Bernatsky S, Hudson M. Antirheumatic drug use and the risk of acute myocardial infarction. Arthrtisis Rheum. 2006;55:531–6.
Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanism of action: a comprehensive review. Pharmacol Ther. 2008;117:244–79.
Nam JL, Winthrop KL, Van Vollenhoven RF, et al. Current evidence for the management of rheumatoid arthritis with biological disease-modifying antirheumatic drugs: a systematic literature review informing the EULAR recommendations for the management of RA. Ann Rheum Dis. 2010;69:976–86.
Pollono EN, Lopez-Olivo MA, Lopez JA, et al. A systematic review of the effect of TNF-alpha antagonists on lipid profiles in patients with rheumatoid arthritis. Clin Rheumatol. 2010;29:947–55.
Van Sijl AM, Peters MJ, Knol DL, et al. The effect on TNF-alpha blocking therapy on lipid levels in rheumatoid arthritis: a meta-analysis. Semin Arthritis Rheum. 2011;41:393–400.
Daien CI, Duny Y, Barnetche T, et al. Effect of TNF inhibitors on lipid profile in rheumatoid arthritis: a systematic review with meta-analysis. Ann Rheum Dis. 2012. doi:10.1136/annrheumdis-2011-201148.
Gasparyan AY, Ayvazyan L, Cocco G, et al. Adverse cardiovascular effects of antirheumatic drugs: implications for clinical practice and research. Curr Pharm Des. 2012;18:1543–55.
Barnabe C, Martin BJ, Ghali WA. Systematic review and metanalysis: anti-tumor necrosis factor alpha therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res. 2011;63:522–9.
Sarzi-Puttini P, Atzeni F, Shoenfeld Y, et al. TNF-alpha, rheumatoid arthritis, and heart failure: rheumatological dilemma. Autoimmun Rev. 2005;4:153–61.
Solomon GE. T-cell agents in the treatment of rheumatoid arthritis. Bull NYU Jt Dis. 2010;68:162–5.
Kerekes G, Soltesz P, Der H, et al. Effects of rituximab treatment on endothelial dysfunction, carotid atherosclerosis, and lipid profile in rheumatoid arthritis. Clin Rheumatol. 2009;59:1821–4.
Gonzalez-Juanatey C, Llorca J, Vazquez-Rodriguez TR, et al. Short-term improvement of endothelial function in rituximab treated rheumatoid arthritis patients refractory to tumor necrosis factor alpha blocker therapy. Arthritis Rheum. 2008;59:1821–4.
Novikova D, Popkova T, Nasonov E. The effect of anti-B-cell therapy on the development of atherosclerosis in patients with rheumatoid arthritis. Curr Pharm Des. 2012;18:1512–8.
Gurcan HM, Keskin DB, Stern JN, et al. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol. 2009;9:10–25.
van Vollenhoven RF, Emery P, Bingham CO, et al. Longterm safety of patients receiving rituximab in rheumatoid arthritis clinical trials. J Rheumatol. 2010;37:558–67.
Papanicolau DA, Wilder RL, Manolagas SC, et al. The pathophysiologic roles of interleukin 6 in human disease. Ann Intern Med. 1998;128:127–37.
Gonzalez-Gay MA, Gonzalez-Juateney C, Lopez-Diaz MJ, et al. HLA-DRB1 and persistent chronic inflammation contribute to cardiovascular events and cardiovascular mortality in patients with rheumatoid arthritis. Arthritis Rheum. 2007;57:125–32.
Poole CD, Conway P, Currie CJ. An evaluation of the association between C-reactive protein, the change in C-reactive protein over one year, and all cause mortality in chronic immune mediated inflammatory disease managed in UK general practice. Rheumatology (Oxford). 2009;48:78–82.
Oldfield V, Dhillon S, Plosket GL. Tocilizumab: a review of its use in the management of rheumatoid arthritis. Drugs. 2009;69:609–32.
Singh JA, Beg S, Lopez-Olivo MA. Tocilizumab for rheumatoid arthritis. Cochrane Database Syst Rev. 2010;7:CD008331.
Nishimoto N, Ito K, Takagi N. Safety and efficacy profiles of tocilizumab monotherapy in Japanese patients with rheumatoid arthritis: meta-analysis of six initial trials and five long term extensions. Md Rheumatol. 2010;20:222–32.
Kawashiri SY, Kawakami A, Yamasaki S, et al. Effects of the anti-interleukin-6 receptor antibody, tocilizumab, on serum lipid levels in patients with rheumatoid arthritis. Rheumatol Int. 2011;31:451–6.
Bisoendial RJ, Stroes ES, Kastelein JJ, et al. Targeting cardiovascular risk in rheumatoid arthritis: a dual role for statins. Nat Rev Rheumatol. 2010;6:157–64.
Vishal T, Bano G, Khajuria V, et al. Pleitropic effects os statins. Ind J Pharmacol. 2003;37:77–85.
Semb AG, Holme I, Kvien TK, et al. Intensive lipid lowering in patients with rheumatoid arthritis and previous myocardial infarction: an explorative analysis from the incremental decrease in endpoints through aggressive lipid lowering (IDEAL) trial. Rheumatology (Oxford). 2011;50:324–9.
De Vera MA, Choi H, Abrahamowicz M, et al. Statin discontinuation and risk of acute myocardial infarction in patients with rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis. 2011;70:1020–4.
Toms TE, Panoulas VF, Douglas KM, et al. Statin use in rheumatoid arthritis in relation to actual cardiovascular risk: evidence for substantial under treatment of lipid-associated cardiovascular risk? Ann Rheum Dis. 2010;69:683–8.
El-Barbary A, Hussein M, Rageh E, et al. Effect of atorvastatin on inflammation and modification of vascular risk factors in rheumatoid arthritis. J Rheumatol. 2011;38:229–35.
McCarey DW, McInnes IB, Madhok R, et al. Trial of atorvastatin in rheumatoid arthritis (TARA): double-blind, randomized placebo-controlled trial. Lancet. 2004;363:2015–21.
Jick SS, Choi H, Li L, et al. Hyperlipidemia, statin use and the risk of developing rheumatoid arthritis. Ann Rheum Dis. 2009;68:546–51.
Winiarska M, Bil J, Wilczek E, et al. Statins impair antitumor effects of rituximab by inducing conformational changes of CD20. PLoS Med. 2008;5:e64.
Arts EE, Jansen TL, Den Broeder A, et al. Statins inhibit the antirheumatic effects of rituximab in rheumatoid arthritis: results from the Dutch Rheumatoid Arthritis Monitoring (DREM) registry. Ann Rheum Dis. 2011;70:877–8.
Peters MJL, Symmons DPM, McCarey D, et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010;69:325–31.
Conroy RM, Pyorala K, Fitzgerald AP, et al. Estimation of 10 year risk of fatal cardiovascular disease in Europe: the SCORE project. Eur Heart J. 2003;24:987–1003.
Chung CP, Oeser A, Avalos I, et al. Utility of the Framingham risk score to predict the presence of coronary atherosclerosis in patients with rheumatoid arthritis. Arthritis Res Ther. 2006;8:R186.
Acknowledgments
Dr. Suarez-Almazor is the recipient of a K24 award from the National Institute on Arthritis, Musculoskeletal and Skin Disorders (NIAMS K24AR053593). The article’s contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIAMS or NIH.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Amezaga Urruela, M., Suarez-Almazor, M.E. Lipid Paradox in Rheumatoid Arthritis: Changes With Rheumatoid Arthritis Therapies. Curr Rheumatol Rep 14, 428–437 (2012). https://doi.org/10.1007/s11926-012-0269-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11926-012-0269-z