
Abstract
Substantial evidence exists that in addition to the well-known complications of diabetes, increased fracture risk is an important morbidity. This risk is probably due, at least in part, to altered bone remodeling and bone cell function in diabetes. Circulating biochemical markers of bone formation, including P1NP, osteocalcin and bone-specific alkaline phosphatase have been found to be decreased in type 2 diabetes (T2D) and may be predictive of fractures independently of bone mineral density (BMD). These findings have been corroborated by preliminary histomorphometric data. Reductions in the bone resorption marker serum CTx in T2D have also been reported. Serum sclerostin levels have been found to be increased in T2D and appear to be predictive of fracture risk independent of BMD. Other factors such as bone marrow fat saturation, advanced glycation endproduct (AGE) accumulation, and microarchitectural changes might also relate to bone cell function and fracture risk in diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is the most common endocrine disorder with a prevalence of approximately 327 million worldwide [1]. Substantial evidence exists that in addition to the well-known complications of diabetes, such as neuropathy, nephropathy, and retinopathy, increased fracture risk is an important morbidity [2, 3]. Individuals with type 1 diabetes (T1D) have a very high risk of hip fracture, approximately six times greater than those without diabetes [2, 3]. Although the fracture risk is not as high in type 2 diabetes (T2D), it is nevertheless increased as well [4, 5]. A meta-analysis of 12 studies reported a relative risk of 1.7 (95 % CI: 1.3–2.2) for hip fracture in both men and women with T2D [2]. The risk of all clinical fractures was also increased, with a summary RR of 1.2 (95 % CI: 1.0–1.5) [2]. Subsequent studies have reported similar results [6, 7], with a direct association between the duration of diabetes and increased fracture risk [8]. Fractures in all diabetic individuals are particularly problematic because they are associated with poor fracture healing, greater morbidity [9, 10], and greater healthcare costs [11].
In T1D, hip and spine BMD are reduced compared with normative reference populations [3] or with healthy controls [12]. As in broader populations, reduced BMD is associated with higher fracture prevalence among those with T1D [13]. In contrast, BMD is generally higher in those with T2D compared to those without [3]. In a meta-analysis, Vestergaard reported an increased Z-score of +0.41 at the spine and +0.27 at the hip associated with T2D [3]. The paradox of higher BMD in association with increased fractures in T2D might be attributed to more frequent trauma, as diabetes is associated with an increased frequency of falls. However, in studies of diabetes and fracture that controlled for fall frequency, diabetes still remained independently associated with increased fracture risk [6, 14]. Thiazolidinedione (TZD) use might also be considered as an explanation, since it has been proposed that these agents divert mesenchymal stem cells from the osteogenic to the adipocytic lineage and are associated with bone loss and increased fracture risk, particularly in women [15]. However, TZD use cannot fully account for the increased risk of fracture observed with diabetes, since most studies included substantial observation time prior to the widespread use of these medications. Rather, it appears that other bone properties, which are undetectable by DXA, are probably contributing to fracture risk in both T1D and T2D.
Altered bone remodeling in diabetes may be one such factor. Bone remodeling depends upon a coordinated sequence of bone resorption by osteoclasts, followed by bone formation by osteoblasts. Diabetes appears to affect the function of both cell types, as well as that of osteocytes, the most abundant cells in bone.
Type 1 Diabetes
Levels of parathyroid hormone (PTH), a key regulator of bone remodeling, are altered with glycemia and diabetes. In healthy subjects, a glucose load leads to a slight decrease in ionized calcium and an increase in PTH, after an initial temporary decrease, as well as a decrease in markers of both bone formation and resorption [16]. In vitro data similarly show that exposure to high glucose levels impairs osteoblast function [17–19]. In T1D, blunted PTH responses have been observed [20]. In vitro data [18] and in vivo studies involving rodent models of T1D [21] indicate that bone formation is consistently impaired, as shown by the expression of osteoblastic transcription factors, including RUNX2, biochemical markers, and histomorphometric indices [22]. An association between T1D and low bone formation in clinical studies has also been shown [23–25]. In the largest histomorphometry study to date, iliac biopsies in 18 otherwise healthy subjects with T1D were compared with those from healthy age- and sex-matched non-diabetic control subjects [26]. Diabetic subjects, when compared to controls, had no significant differences in mineral apposition rate (MAR), mineralizing surface (MS/BS), osteoid maturation time (Omt), mineralizing osteoid (MS/OS), mineralization lag time (Mlt), bone formation rate (BFR/BS or BFR/BV), formation period (FP), remodeling period (Rm.P), or activation frequency (Ac.F) [26]. It is possible that the clinical data, in contrast to rodent models, showed no difference in bone formation rate for T1D as compared to controls because reduced bone formation emerges only with other complications, such as nephropathy and peripheral neuropathy, and these T1D subjects were otherwise healthy. In a subset of diabetic patients who had fractured, dynamic variables such as BFR/BS, BFR/BV, and Ac.F tended to be lower in the fracturing subjects, perhaps indicating lower remodeling in those T1D subjects [26].
Type 2 Diabetes
PTH and Biochemical Markers of Bone Turnover in T2D
Decreased bone remodeling in T2D has been demonstrated by a number of lines of evidence. Levels of PTH tend to be 20–50 % lower in T2D subjects than in controls, even in the setting of reduced eGFR, suggesting a state of reduced PTH secretion in T2D [27–29]. Circulating biochemical markers of bone formation, including P1NP, osteocalcin [28, 29], and bone-specific alkaline phosphatase [30], have been found to be decreased in T2D. These decreases in formation measures are associated with reductions in the bone resorption marker serum CTx [27–30]. The decrease in bone remodeling in T2D appears to be predictive of fracture risk regardless of BMD. In a study of 255 T2D women and 240 controls, T2D women with the combination of the lowest PTH and osteocalcin levels had nearly a fivefold increased risk of vertebral fractures independent of lumbar spine BMD [29].
Dynamic Histomorphometry in T2D
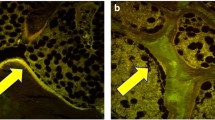
Lower bone formation in T2D on biopsy was reported in one study, but the numbers were very small (n = 6 T2D patients; 2 female), and the results were confounded by selecting for low BMD and a problematical control group [31]. In a more recent pilot study, low bone formation was observed in six T2D postmenopausal women as compared to six postmenopausal age-matched non-diabetic controls, where tetracycline double-labeled iliac crest bone biopsies showed virtually no uptake of label in diabetic subjects (Fig. 1), with reduced mineralizing surface, osteoid surface, and osteoblast surface (Fig. 2) [32]. These preliminary histological data corroborate the decrease in biochemical markers of bone turnover.

Histomorphometric changes in bone formation. Tetracycline double-labeled bone biopsies in a 58-yr-old T2D Caucasian woman (a) and a 57-yr-old Caucasian female control (b). Bone formation is decreased in T2D with reduced mineralizing surface. The arrows highlight tetracycline uptake in the control subject and the absence of uptake in the diabetic subject. Adapted with permission from [32]

Quantitative measures of bone formation were lower in T2D postmenopausal women than in controls. Adapted with permission from [32]
Other Bone Markers in T2D
IGF-1, an anabolic factor which stimulates osteoblast proliferation, has been inversely associated with the risk and number of vertebral fractures in diabetic women independent of BMD [28, 33]. Another marker which might reflect bone formation is that of circulating osteogenic precursor (COP) cells [34], which have been reported to be decreased in patients with T2D. COP cells can be detected in the peripheral blood by flow cytometry using antibodies specific for the osteoblast matrix protein osteocalcin (OCN) [35]. Peripheral blood mononuclear cells that were positive for osteocalcin were lower in postmenopausal women with T2D as compared to non-diabetic controls [32]. Moreover, within the decreased pool of overall OCN+ cells, the T2D subjects had an increased subpopulation of immature OCN+ cells, i.e., cells that also had early markers CD146 and CD34, subpopulations which diminish when osteoblasts mature [34]. An additional novel bone marker in T2D may be sphingosine 1-phosphate (S1P), a lipid mediator which increases osteoclastogenesis by increasing RANKL [36]. S1P was found to be increased in T2D women (n = 482) as compared to controls and was associated with vertebral fractures. Interestingly, this marker suggests an elevation in bone resorption in T2D, in contrast to the reports of decreased s-CTx levels [27–30].
Sclerostin in T2D
Sclerostin, an osteocyte product, is a negative regulator of bone formation which competes with the anabolic Wnt β-catenin pathway by binding to LRP5 or 6 [37]. In healthy adults, sclerostin levels are increased by factors including age, BMI, inactivity, bone mineral content, and possibly fractures [37]. It was first reported in 2012 that sclerostin levels were higher in 74 T2D women and men vs. 50 non-diabetic controls and that higher levels correlated with age, male gender, and BMD [38]. This observation was corroborated by another report in which sclerostin levels were found to be twofold higher in T2D than in controls or T1D, after adjusting for age and BMI [39]. A correlation between Wnt disruption and decreased osteoblast activity was further observed in 40 T2D postmenopausal women who, as compared to controls, had decreased β-catenin levels which correlated with lower BAP [30]. In the largest diabetes sclerostin study, higher sclerostin levels in 321 men and women with T2D were associated with an increased risk of vertebral fractures independent of lumbar spine BMD [40]. It could be posited from these data that the higher sclerostin levels in T2D reflect the presence of more deeply embedded osteocytes in older bone that has accumulated more microscopic damage.
Bone Marrow Fat Saturation in T2D
Recent data suggest that altered bone marrow fat composition is linked with diabetic fragility fractures independent of BMD [41]. In a study of 69 postmenopausal women, MR spectroscopy (MRS) of the lumbar spine quantified vertebral bone marrow fat content and composition in diabetic and non-diabetic subjects with and without fragility fractures. After adjustment for age, race, and spinal vBMD, diabetes was associated with −1.3 % (CI −2.3 to −0.2 %, p = 0.018) lower unsaturation and +3.3 % (CI 1.1 to 5.4 %, p = 0.004) higher saturation levels, with fractured diabetics having the lowest marrow unsaturation and highest saturation. It is conceivable that this type of alteration in bone marrow fat might reflect a shift in stem cell lineage away from osteoblastogenesis toward adipogenesis [42]. In the AGES-Reykjavik cohort (115 men and 134 women; mean age 79; 7 % with diabetes), sclerostin levels were positively associated with marrow fat by MRS in men independent of BMD [42]. This positive relationship could be explained by a shift in precursor stem cell lineage away from osteoblastogenesis, as reflected by higher sclerostin levels, toward adipogenesis, as reflected by higher bone marrow fat [42].
AGEs and Bone Remodeling
Decreased bone formation might occur in part because of increased advanced glycation endproducts (AGEs) in bone collagen. In the setting of chronic hyperglycemia, AGEs accumulate in the organic bone matrix by a process known as non-enzymatic glycation (the Maillard reaction) [43–50]. AGEs interfere with normal osteoblast function [51] and attachment to the collagen matrix [52], as well as impair osteoblast development [53, 54]. Moreover, low bone formation also works in the opposite direction to further increase AGEs, as for example with high bisphosphonate dosages [44]. Accumulation of AGEs in the organic bone matrix leads to more biomechanically brittle bone that has lost its toughness and is less able to deform before fracturing [44]. Urinary pentosidine, the best studied AGE, was associated with a 42 % increase in clinical fracture incidence in T2D [55]. AGEs thus appear to be related to both low bone formation and increased bone fragility in T2D.
Architectural Properties in T2D
In addition to alterations in bone cells and remodeling, another factor that may contribute to the paradox of increased fractures despite normal areal BMD in T2D is microarchitectural abnormality. Increased cortical porosity, a key determinant of bone fragility [56], has been reported at the radius and tibia in female diabetics who have fractured, as measured by intra-cortical pore volume fraction via high-resolution peripheral quantitative computed tomography (HR-pQCT) [57]. In a recent community-based study of women and men, T2D and increased HbA1c levels were associated with deficits in cortical microstructure and density at the distal tibia [58]. Marcroarchitectural deficits in bone geometry might also explain reduced bone strength in T2D. Strength-to-load ratios (QCT) at the spine and femoral neck were not improved in older adults with T2D although areal BMD (DXA) was higher [59]. In a study of older men, volumetric BMD (pQCT) was higher but bone area was smaller at the distal radius and tibia [60]. Smaller cross-sectional area suggests that stimulation of periosteal apposition, normally observed with greater loading, may be reduced in diabetes. These data seem to suggest that the higher areal BMD in diabetics does not result in biomechanical advantage.
Conclusions
At a time when classical complications of diabetes mellitus are becoming less common due to improved glucose control, the skeleton has emerged as a target organ for disease complications. Alterations in diabetic bone metabolism, including bone cells, remodeling, and other properties, could potentially contribute to this increase in fracture risk. Further investigation of skeletal parameters would shed light on the issue of greater bone fragility in diabetes and potentially offset serious challenges in this population as they age.
References
IDF Diabetes Atlas Update 2012. http://www.idf.org/diabetesatlas/5e/Update2012 (accessed January 10, 2013).
Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol. 2007;166(5):495–505.
Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—a meta-analysis. Osteoporos Int. 2007;18(4):427–44.
Schwartz AV, Vittinghoff E, Bauer DC, et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. JAMA : J Am Med Assoc. 2011;305(21):2184–92.
Giangregorio LM, Leslie WD, Lix LM, et al. FRAX underestimates fracture risk in patients with diabetes. J Bone Miner Res. 2012;27(2):301–8.
Bonds DE, Larson JC, Schwartz AV, et al. Risk of fracture in women with type 2 diabetes: the Women's Health Initiative Observational Study. J Clin EndocrinolMetabol. 2006;91(9):3404–10.
Leslie WD, Lix LM, Prior HJ, Derksen S, Metge C, O'Neil J. Biphasic fracture risk in diabetes: a population-based study. Bone. 2007;40(6):1595–601.
de Liefde II, van der Klift M, de Laet CE, van Daele PL, Hofman A, Pols HA. Bone mineral density and fracture risk in type-2 diabetes mellitus: the Rotterdam Study. Osteoporos Int. 2005;16(12):1713–20.
Hernandez RK, Do TP, Critchlow CW, Dent RE, Jick SS. Patient-related risk factors for fracture-healing complications in the United Kingdom General Practice Research database. Acta Orthop. 2012;83(6):653–60.
Semel J, Gray JM, Ahn HJ, Nasr H, Chen JJ. Predictors of outcome following hip fracture rehabilitation. PM & R : J Injury, Function, Rehabilitation. 2010;2(9):799–805.
Bass E, French DD, Bradham DD. A national perspective of Medicare expenditures for elderly veterans with hip fractures. J Am Med Directors Assoc. 2008;9(2):114–9.
Pan H, Wu N, Yang T, He W. Association between bone mineral density and type 1 diabetes mellitus: a meta-analysis of cross-sectional studies. Diabetes Metab Res Rev. 2014;30(7):531–42.
Neumann T, Samann A, Lodes S, et al. Glycaemic control is positively associated with prevalent fractures but not with bone mineral density in patients with type 1 diabetes. Diabetic, Medicine: J British Diabetic Assoc. 2011;28(7):872–5.
Schwartz AV, Sellmeyer DE, Ensrud KE, et al. Older women with diabetes have an increased risk of fracture: a prospective study. J Clin Endocrinol Metabolism. 2001;86(1):32–8.
Loke YK, Singh S, Furberg CD. Long-term use of thiazolidinediones and fractures in type 2 diabetes: a meta-analysis. CMAJ. 2009;180(1):32–9.
Clowes JA, Allen HC, Prentis DM, Eastell R, Blumsohn A. Octreotide abolishes the acute decrease in bone turnover in response to oral glucose. J Clin Endocrinol Metabolism. 2003;88(10):4867–73.
Inaba M, Nishizawa Y, Mita K, et al. Poor glycemic control impairs the response of biochemical parameters of bone formation and resorption to exogenous 1,25-dihydroxyvitamin D3 in patients with type 2 diabetes. Osteoporos Int. 1999;9(6):525–31.
Inaba M, Terada M, Koyama H, et al. Influence of high glucose on 1,25-dihydroxyvitamin D3-induced effect on human osteoblast-like MG-63 cells. J Bone Miner Res. 1995;10(7):1050–6.
Terada M, Inaba M, Yano Y, et al. Growth-inhibitory effect of a high glucose concentration on osteoblast-like cells. Bone. 1998;22(1):17–23.
Schwarz P, Sorensen HA, Momsen G, Friis T, Transbol I, McNair P. Hypocalcemia and parathyroid hormone responsiveness in diabetes mellitus: a tri-sodium-citrate clamp study. Acta Endocrinol (Copenh). 1992;126(3):260–3.
Botolin S, Faugere MC, Malluche H, Orth M, Meyer R, McCabe LR. Increased bone adiposity and peroxisomal proliferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology. 2005;146(8):3622–31.
McCabe L, Zhang J, Raehtz S. Understanding the skeletal pathology of type 1 and 2 diabetes mellitus. Crit Rev Eukaryot Gene Expr. 2011;21(2):187–206.
Gunczler P, Lanes R, Paoli M, Martinis R, Villaroel O, Weisinger JR. Decreased bone mineral density and bone formation markers shortly after diagnosis of clinical type 1 diabetes mellitus. J Pediatric End Ocrinol Metabolism : JPEM. 2001;14(5):525–8.
Gunczler P, Lanes R, Paz-Martinez V, et al. Decreased lumbar spine bone mass and low bone turnover in children and adolescents with insulin dependent diabetes mellitus followed longitudinally. J Pediatric Endocrinol Metabolism : JPEM. 1998;11(3):413–9.
Masse PG, Pacifique MB, Tranchant CC, et al. Bone metabolic abnormalities associated with well-controlled type 1 diabetes (IDDM) in young adult women: a disease complication often ignored or neglected. J Am Coll Nutr. 2010;29(4):419–29.
Armas LA, Akhter MP, Drincic A, Recker RR. Trabecular bone histomorphometry in humans with type 1 diabetes mellitus. Bone. 2012;50(1):91–6.
Dobnig H, Piswanger-Solkner JC, Roth M, et al. Type 2 diabetes mellitus in nursing home patients: effects on bone turnover, bone mass, and fracture risk. J Clin Endocrinol Metab. 2006;91(9):3355–63.
Ardawi MS, Akhbar DH, Alshaikh A, et al. Increased serum sclerostin and decreased serum IGF-1 are associated with vertebral fractures among postmenopausal women with type-2 diabetes. Bone. 2013;56(2):355–62.
Yamamoto M, Yamaguchi T, Nawata K, Yamauchi M, Sugimoto T. Decreased PTH levels accompanied by low bone formation are associated with vertebral fractures in postmenopausal women with type 2 diabetes. J Clin Endocrinol Metab. 2012;97(4):1277–84.
Gaudio A, Privitera F, Battaglia K, et al. Sclerostin levels associated with inhibition of the Wnt/beta-catenin signaling and reduced bone turnover in type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97(10):3744–50.
Krakauer JC, McKenna MJ, Buderer NF, Rao DS, Whitehouse FW, Parfitt AM. Bone loss and bone turnover in diabetes. Diabetes. 1995;44(7):775–82.
Manavalan JS, Cremers S, Dempster DW, et al. Circulating osteogenic precursor cells in type 2 diabetes mellitus. J Clin Endocrinol Metabolism. 2012;97(9):3240–50.
Kanazawa I, Yamaguchi T, Sugimoto T. Serum insulin-like growth factor-I is a marker for assessing the severity of vertebral fractures in postmenopausal women with type 2 diabetes mellitus. Osteoporos Int. 2011;22(4):1191–8.
Eghbali-Fatourechi GZ, Modder UI, Charatcharoenwitthaya N, et al. Characterization of circulating osteoblast lineage cells in humans. Bone. 2007;40(5):1370–7.
Rubin MR, Manavalan JS, Dempster DW, et al. Parathyroid hormone stimulates circulating osteogenic cells in hypoparathyroidism. J Clin Endocrinol Metabolism. 2011;96(1):176–86.
Ardawi M, Akbar D, Rouzi A, Senani N, Ahmad A, Qari M. Elevated sphingosine 1-phosphate levels are associated with vertebral fractures in patients with type 2 diabetes mellitus. J Bone Miner Res, 2014. http://www.asbmr.org/education/AbstractDetail?aid=51ef5a65-8072-4369-b2b7-c1381053da59 (accessed December 11, 2014).
Canalis E. Wnt signalling in osteoporosis: mechanisms and novel therapeutic approaches. Nat Rev Endocrinol. 2013;9(10):575–83.
Garcia-Martin A, Rozas-Moreno P, Reyes-Garcia R, et al. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97(1):234–41.
Gennari L, Merlotti D, Valenti R, et al. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J Clin Endocrinol Metab. 2012;97(5):1737–44.
Yamamoto M, Yamauchi M, Sugimoto T. Elevated sclerostin levels are associated with vertebral fractures in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2013;98(10):4030–7.
Patsch JM, Li X, Baum T, et al. Bone marrow fat composition as a novel imaging biomarker in postmenopausal women with prevalent fragility fractures. J Bone Miner Res 2013.
Ma YH, Schwartz AV, Sigurdsson S, et al. Circulating sclerostin associated with vertebral bone marrow fat in older men but not women. J Clin Endocrinol Metab. 2014;99(12):E2584–90.
Saito M, Mori S, Mashiba T, Komatsubara S, Marumo K. Collagen maturity, glycation induced-pentosidine, and mineralization are increased following 3-year treatment with incadronate in dogs. Osteoporos Int. 2008;19(9):1343–54.
Tang SY, Allen MR, Phipps R, Burr DB, Vashishth D. Changes in non-enzymatic glycation and its association with altered mechanical properties following 1-year treatment with risedronate or alendronate. Osteoporos Int. 2009;20(6):887–94.
Vashishth D. The role of the collagen matrix in skeletal fragility. Curr Osteoporosis Reports. 2007;5(2):62–6.
Garnero P, Borel O, Gineyts E, et al. Extracellular post-translational modifications of collagen are major determinants of biomechanical properties of fetal bovine cortical bone. Bone. 2006;38(3):300–9.
Schalkwijk CG, Stehouwer CD, van Hinsbergh VW. Fructose-mediated non-enzymatic glycation: sweet coupling or bad modification. Diabetes Metab Res Rev. 2004;20(5):369–82.
Monnier VM, Kohn RR, Cerami A. Accelerated age-related browning of human collagen in diabetes mellitus. Proc Natl Acad Sci U S A. 1984;81(2):583–7.
Schwartz AV. Diabetes mellitus: does it affect bone? Calcif Tissue Int. 2003;73(6):515–9.
Reiser KM. Nonenzymatic glycation of collagen in aging and diabetes. Proc Soc Exp Biol Med. 1998;218(1):23–37.
Sanguineti R, Storace D, Monacelli F, Federici A, Odetti P. Pentosidine effects on human osteoblasts in vitro. Ann N Y Acad Sci. 2008;1126:166–72.
McCarthy AD, Uemura T, Etcheverry SB, Cortizo AM. Advanced glycation endproducts interefere with integrin-mediated osteoblastic attachment to a type-I collagen matrix. Int J Biochem Cell Biol. 2004;36(5):840–8.
McCarthy AD, Etcheverry SB, Cortizo AM. Effect of advanced glycation endproducts on the secretion of insulin-like growth factor-I and its binding proteins: role in osteoblast development. Acta Diabetol. 2001;38(3):113–22.
Kume S, Kato S, Yamagishi S, et al. Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage, and bone. J Bone Miner Res. 2005;20(9):1647–58.
Schwartz AV, Garnero P, Hillier TA, et al. Pentosidine and increased fracture risk in older adults with type 2 diabetes. J Clin Endocrinol Metab. 2009;94(7):2380–6.
Schaffler MB, Burr DB. Stiffness of compact bone: effects of porosity and density. J Biomech. 1988;21(1):13–6.
Patsch JM, Burghardt AJ, Yap SP, et al. Increased cortical porosity in type 2 diabetic postmenopausal women with fragility fractures. J Bone Miner Res. 2013;28(2):313–24.
Samelson E, Bouxsein M, Brochin E, et al. Deficits in cortical bone density and microstructure in type 2 diabetes: Framingham HR-pQCT Study. J Bone Miner Res, 2014. http://www.asbmr.org/education/AbstractDetail?aid=717f3c4f-ac78-4141-be67-445acf5d4c9e (accessed December 12, 2014).
Melton 3rd LJ, Riggs BL, Leibson CL, et al. A bone structural basis for fracture risk in diabetes. J Clin Endocrinol Metabol. 2008;93(12):4804–9.
Petit MA, Paudel ML, Taylor BC, et al. Bone mass and strength in older men with type 2 diabetes: the Osteoporotic Fractures in Men Study. J Bone Miner Res. 2010;25(2):285–91.00.
Funding Source
Columbia University Irving Institute for Clinical and Translational Research CTSA/CTO pilot award.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
MR Rubin declares no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by MR Rubin involving animal and/or human subjects were performed after approval by the appropriate institutional review boards. When required, written informed consent was obtained from all participants.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Bone and Diabetes
Rights and permissions
About this article

Cite this article
Rubin, M.R. Bone Cells and Bone Turnover in Diabetes Mellitus. Curr Osteoporos Rep 13, 186–191 (2015). https://doi.org/10.1007/s11914-015-0265-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-015-0265-0