
Abstract
Bone mineral density alone cannot reliably predict fracture risk in humans and laboratory animals. Therefore, other factors including the quality of organic bone matrix components and their interactions may be of crucial importance to understanding of fragility fractures. Emerging research evidence shows, that in addition to collagen, certain noncollagenous proteins (NCPs) play a significant role in the structural organization of bone and influence its mechanical properties. However, their contribution to bone strength still remains largely undefined. Collagen and NCPs undergo different post-translational modifications, which alter the quality of the extracellular matrix and the response of bone to mechanical load. The primary focus of this overview is on NCPs that, together with collagen, contribute to structural and mechanical properties of bone. Current information on several mechanisms through which some NCPs influence bone’s resistance to fracture, including the role of nonenzymatic glycation, is also presented.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The molecular origin of the remarkable mechanical properties of bone is still not fully understood. Over the lifetime of vertebrates, mechanical properties deteriorate and lead to weaker bones that are prone to fracture. Bone is a hierarchically structured nanocomposite consisting of mineral (hydroxyapatite) and organic matrix. The organic matrix is composed of fibrillar (primarily collagen I) and a variety of nonfibrillar proteins [1]. Traditionally, the inorganic hydroxyapatite part has been considered to be the most important factor influencing the mechanical properties of bone, because it provides stiffness and strength to bone. To date, the amount of mineral in a patient is used worldwide to predict fracture risk and to assess response to therapy. However, despite the association between low bone mineral density (BMD) and high risk of fracture, there is an overlap in BMD between fracture and non-fracture patients [2, 3]. Additionally, a small fraction of patients undergoing long-term bisphosphonate treatment show an increased risk for femoral shaft fractures despite an increase in BMD [4, 5, 6•].
The assessment of BMD relies primarily on dual-energy X-ray absorptiometry (DEXA), a technique that provides limited information on bone structure and matrix. Other imaging modalities such as high-resolution peripheral quantitative computed tomography (HR-pQCT) and magnetic resonance (MR) offer characterization of bone architecture, but their cost and inaccessibility have precluded them from becoming widely used [7]. None of the aforementioned techniques are able to monitor the variations in bone matrix quality, which result from the alterations of the bone’s organic matrix, and these variations are known to be the major contributors of post-yield deformation and fracture [8].
Importantly, the deterioration of bone quality resulting in more fragile tissue cannot be attributed solely to one component of bone, but rather to the changes that occur in the mineral and collagen as well as alterations to noncollagenous proteins (NCPs). NCPs regulate the matrix assembly and vary during aging, disease, and anti-osteoporosis treatments [9–11]. These undesired changes tend to increase fracture risk. Often, low bone mass leads to fragility fractures; however, many patients diagnosed with low bone mass do not experience severe fractures [12]. Thus, it is important to take into account the organic portion of bone to understand the complex changes in bone that occur at macro-, micro-, and nanoscale levels, and to determine what parameters contribute to the decreased bone quality of osteoporotic tissue. Such new information will enable the development of new diagnostic tools and more effective therapies.
Extracellular Bone Matrix
Collagen is the most abundant component of the extracellular bone matrix. In addition to collagen, bone matrix contains approximately 180 to 200 NCPs. In a simplistic view, NCPs can be divided into two major groups. One group plays a primarily structural and mechanical role (Table 1) and the other modulates function of different bone cells by interacting with their cell-surface receptors, proteases, hormones, and other biomolecules including proteoglycans and collagens (Table 2) [13]. Notably, some proteins may play multiple roles and can be categorized into either group (eg, osteocalcin [OC] or osteopontin [OPN]). A variety of NCPs [1, 13], including fibronectin, OC, OPN, osteonectin, bone sialoprotein II, decorin, and biglycan, are present in a substantial amount (0.1% to 2.0%). Of this group, only OC and bone sialoprotein II are specific to bone. Other NCPs are also present in a number of noncalcified tissues.
The second group of NCPs includes growth factors, and many of them are stored in the bone matrix [14]. Growth factors regulate distinct cellular processes including bone cell proliferation, differentiation, and matrix remodeling of bone. Examples include transforming growth factors-β (TGF-β1, TGF-β2, and TGF-β3) [15], insulin-like growth factors [16], and bone morphogenic proteins (BMPs) [17•].
Members of the two NCP groups described above interact with each other and with collagens at different levels such as regulation of cellular gene expression and protein activity. For example, bone sialoprotein II and OPN increase intracellular calcium in osteoclasts [18]. Decorin has been shown to bind to TGF-β and enhance its activity [19]. TGF-β modulates OPN and OC production [14, 20]. A variety of NCPs interacts with collagen and regulate bone mineralization. The role of NCPs in mineralization is discussed in the next section after a brief overview of collagens and collagen type I fibrils.
Collagens
Collagen types, their amount, and distribution in bone vary during the development and growth of an adult skeleton. In addition to collagen type I, developing fetal bones contain much higher levels of collagen III, V, and VI compared to bones of children and adults [21, 22]. For example, in fetal bone, type VI collagen is present in discrete fibrils separate from type III collagen, and by the age of 7 years, it becomes restricted to the margins of bone cells and bone surfaces [21]. In adults, collagen type I represents about 90% of the total protein matrix and it forms fibrils that are 80 to 100 nm in diameter and 10 μm or more in length. The fully assembled three-dimensional fibrils have a regular series of gaps and channels. Mineral deposits occur within these gaps and channels [23•, 24•]. The pore-hole combination provides space for growth of plate-like crystals transversely through the collagen fibrils. Therefore, the nucleation of mineralization in bone is also quite specific to the collagen type. Several unique NCPs have been shown to strongly associate with the mineralized collagen and contribute toward the quality of the filamentous scaffold. Some of these proteins are central to this overview and will be discussed in more detail in the following sections.
Collagens also interact with many other proteins and peptides in the extracellular matrix that do not directly contribute to the nucleation and growth of crystals. Such proteins include fibronectin [25], laminins [26, 27], and proteoglycans [28], as well as some cell-surface receptors such as integrins [29]. Proteoglycans play an important role in organizing the bone extracellular matrix and take part in the structuring of the tissue as active regulators of collagen fibrillogenesis. The most abundant modification of proteoglycans is heparin. Type V collagen possesses a site that binds heparin/heparin sulfate under physiological conditions [30]. Proteoglycans also display selective patterns of reactivity with cytokines and growth factors, such as TGF-β or osteoprotegerin [14] and, thus, modulate the availability and biological activity of these proteins in bone.
Growth factors and hormones form very large and diverse superfamilies that play a central role in cellular growth, differentiation, survival, and cell cycle progression in different tissues. They also have a profound effect on bone remodeling processes. In this overview, only selected studies will be discussed to illustrate recent research developments, with a specific focus on the TGF-β superfamily and its role in osteoporosis and bone fragility. A number of excellent reviews are available and recommended for readers interested in a broad, in-depth survey of other growth factors [15, 16, 17•, 31] and growth factor-related proteins [16, 32].
Transforming Growth Factor-β
The TGF-β superfamily is an evolutionary conserved family of structurally related dimeric cytokines with representatives in organisms as diverse as mammals and invertebrates. The superfamily includes TGF-βs, BMPs [17•], growth and differentiation factors, activins [33], inhibin [34], and anti-Müllerian hormone [35, 36]. TGF-β1 is the most abundant isoform among TGF-βs. The largest sources of TGF-β1 are platelets (20 mg/kg) [37] and bone (200 μg/kg) [38]. All three isoforms of TGF-βs (TGF-β1, -β2, and -β3) are detected in bone. The TGF-β1 isoform is the most abundant at the protein level [39]. Members of the TGF-β family are secreted as latent complexes. Thus, TGF-β latent complexes need to be activated to exhibit biological activity. Once activated, TGF-βs can interact with their receptors to induce signaling [40]. The Smad proteins play a central role in the transmission of signals from all receptors activated by the TGF-β superfamily members to target genes in the nucleus [41, 42].
Mouse models of increased (D4 and D5 mice) and decreased (DNTβRII, Smad3+/− and Smad3−/− mice) TGF-β signaling have been generated and mechanically tested to investigate their role in determining the composition of bone matrix and fragility [43]. Using nanoindentation in combination with atomic force microscopy, Balooch et al. [43] established that TGF-β regulates elastic (Young’s) modulus and hardness of bone matrix. Mice tibias with increased TGF-β levels (D4 and D5 mice bones) displayed considerable heterogeneity in the local elastic modulus compared to wild-type mice bones. These mice also showed decreased mineral concentration throughout the tibia. Conversely, reduced TGF-β signaling in DNTβRII or Smad3−/− mice led to increased mineral concentration. As a result, decreased levels of TGF-β increased elastic modulus and hardness of bone matrix by 33% in DNTβRII mice and up to 42% in Smad3−/− mice compared to wild-type controls.
To investigate the role of TGF-β signaling in the macromechanical properties of bone, Balooch et al. [43] subjected mice femurs to a 3-point bending test. Femurs from D4 and D5 mice showed a 31% or 29% decrease in fracture toughness, respectively, relative to wild-type bones. Conversely, bones from DNTβRII or Smad3+/− mice with decreased TGF-β responsiveness exhibited 43% and 49% higher fracture toughness compared to the wild-type controls [44]. Consistent with its decreased bone mass and bone mineral concentration, 2-month-old Smad3−/− bone had a 30% lower resistance to fracture than the wild-type controls. Data collected by Balooch et al. [43] also showed that TGF-β signaling regulates the mechanical properties of bone matrix independently of changes in bone mass and architecture. This seminal work is yet another indication that bone matrix proteins are critical determinants of material properties of the bone and the variations in their amount and quality should be carefully investigated when determining the efficacy of drug treatments on bone quality.
Contributions of NCPs to the Mechanical Properties of Bone
The contributions of noncollagenous components of the extracellular matrix to bone strength are largely undefined and their roles are mainly limited to non-architectural elements of bone matrix [43, 45, 46]. NCPs comprise a total of approximately 10% of the organic matrix [47]. A number of NCPs interacts with collagen fibrils and may function as “glue” enhancing bone’s resistance to fracture. NCPs also facilitate crystal nucleation, crystal growth, shape, and orientation [48, 49] and, thus, are involved in the formation of the fundamental building blocks of bone tissue, the mineralized collagen fibrils. A better insight into the role of certain NCPs has been achieved recently and these results are discussed in the following sections.
Osteopontin
OPN is one of the most abundant NCPs in bone matrix (1% to 2% out of total 10% NCPs in a healthy bone). This multifunctional protein is highly expressed in bone and many other organs. It is involved in both physiological and pathological processes including but not limited to biomineralization, inflammation, leukocyte recruitment, and cell survival [50]. While most of the noncollagenous bone matrix proteins are nearly homogeneously distributed throughout bone, particularly high levels of OPN are observed at cement lines in remodeling bone and at the laminae limitantes on bone surfaces [51]. Thus, it came as a true surprise when mice lacking OPN (OPN(−/−)) showed normal development and bone structure [52]. Since the evidence prior to generation of OPN(−/−) mouse indicated the involvement of OPN in the calcification of skeletal tissues, Rittling et al. [52] proposed that hard tissues can utilize alternative calcification strategies, which do not involve OPN. Consequently, a variety of anionic proteins have been identified as regulators of calcification in vertebrates and invertebrates.
Recent work from the field of biomechanics, specifically from testing of bone’s resistance to fracture, has brought additional new and interesting insights into the biomechanical role of OPN in bone tissue. When bone fails due to tensile or compressive mechanical overload, the main fracture mechanism appears to be microcracking resulting from the delamination of mineralized collagen fibrils [53]. Fantner et al. [54] demonstrated the role of Ca2+-ion-mediated networks in the energy dissipation by bone. Moreover, these authors were able to link the energy dissipation effects in bone to NCPs, specifically OPN [55]. In the same work, it was also established that OPN functions as glue holding the bone matrix together. The authors proposed that a nonfibrillar protein such as OPN may act as glue to prevent the separation of the mineralized collagen fibrils, and thereby counteract the formation of cracks. This protective effect relies on energy dissipation through the rupture of weak intramolecular bonds of the glue protein and its stretching. Intramolecular bonds are weak, but they exist in large numbers in the extracellular bone matrix due to the relatively high amount of OPN in bone (0.1% to 0.2%) and its large size (∼62 kDa). Consequently, the external energy needed to break all weak bonds and fracture bone is large. After removal of the applied force the weak bonds (termed “sacrificial bonds”) can undergo complete or partial restoration, and this behavior allows bone to regain some of its lost strength. How all the above knowledge can be translated to possibly reverse the effects of osteoporosis remains an open question.
Our recent work on the biochemical analysis of younger (osteonal) and older (interstitial) bone tissue revealed a three- to fourfold decline in the amount of OPN in bone due to an increase in tissue age [56••]. Less OPN implies less glue to prevent separation of collagen fibers under the applied mechanical force and less energy to rupture OPN’s sacrificial bonds. It is also known, that at the structural level, OPN interacts with collagen, OC, and other NCPs. Decline in the OPN amount in bone will impact such interactions and may lead to diminished quality of bone due to matrix-based differences in collagen mineralization that are observed between normal and aged tissues [57•].
Osteocalcin
OC is the most abundant NCP of bone extracellular matrix and is synthesized exclusively by osteoblasts [58, 59]. This multifunctional protein is known as a regulator of the hydroxyapatite crystals’ growth and it controls the size of growing crystals in a concentration-dependent manner without impairing bone resorption or mineralization. OC-deficient (OC−/−) mice developed by Ducy et al. [60] were normal at birth and showed no skeletal defects. However, as opposed to OPN(−/−) mice, over time, the OC(−/−) mice developed abnormalities in bone remodeling, which became clearly noticeable at 6 months of age. Specifically, the long bones from OC(−/−) mice had increased cortical thickness and density compared to the wild-type littermates. The 9-month-old animals showed additional increase of the width of the diaphysis and had more cancellous bone than the wild-type littermates. These changes suggest that the lack of OC may lead to increased mineral acquisition.
Recently, using the proteomics approach, we determined that younger osteonal bone tissue contains significantly higher OC (also OPN, as discussed above) levels than older interstitial bone [56••]. In particular, there was 20-fold higher content of OC in osteonal bone compared to interstitial bone. Since both OPN and OC have been shown to regulate bone remodeling [61], the observed differences reveal unexpected changes in bone matrix composition during aging. In this context, it would be important to determine how mouse OC(−/−) bones differ from OPN(−/−) bones when mechanically tested.
Osteonectin
Osteonectin, also known as SPARC (secreted protein acidic and rich in cysteine), is a multifunctional, 32- to 50-kDa glycoprotein (the determined molecular size differs between organisms and conditions used for gel separation) that is expressed in many different tissues, including bone [62]. As a multifunctional protein, osteonectin supports bone remodeling [63], regulates cell proliferation and cell–matrix interactions [64, 65], and stimulates angiogenesis and the production of matrix metalloproteinases, including gelatinase, stromelysin, and collagenase-1 [66, 67]. In vitro studies show the osteonectin-collagen type I complex binds synthetic apatite crystals and the available free calcium ions. The complex also nucleates mineral phase deposition in vitro from metastable balanced salt solutions [62]. Antibodies raised against osteonectin cross-react with bone and, to a lesser extent, with dentin [68]. In bone, the protein is localized to mineralized trabeculae and is present in a higher amount in the matrix than in the cells of bone [63]. Some studies suggest that, in bone, osteonectin (ONT) links the mineral and collagen, perhaps by initiating active mineralization in normal skeletal tissue [62, 69].
Mice with a homozygous-null mutation in the osteonectin gene are born with no obvious abnormalities [70, 71]. However, as early as at 1.5 months of age they undergo progressive early-onset cataractogenesis. Subsequent comprehensive study of the skeleton of the ONT(−/−) mice revealed that the ONT-null mutation severely affects trabecular bone present in vertebrae and the metaphysis of long bones [60]. Particularly detrimental was the effect of ONT-null mutation on trabecular bone microarchitecture (60% fewer trabeculae than the age-matched control animals). The trabecular bone volume in mutant mice was approximately 50% lower than in control mice. Decreased radiographic density and decreased trabecular bone were also evident in the metaphyseal and epiphyseal regions of the tibia and femur. It is noteworthy that trabecular bone is responsible for much of the metabolic function of bone and it is most readily lost during calcium deficiency, estrogen deficiency, or loss of weight bearing [72, 73]. The ONT(−/−) mice did not show significant difference in cortical bone thickness at any age [60]. The differential effect of the ONT-null mutation on trabecular and cortical bone may depend on biochemical differences and distinct regulatory mechanisms between these bone tissues.
Biomechanical studies of bones from ONT(−/−) animals assayed through a 3-point bending test revealed that, beginning at 17 weeks of age, there was some decrease in bone strength (maximum load resistance) and bone flexibility (stiffness). Since several bone matrix proteins, including OC and OPN, are produced at a similar level in ONT(−/−) mouse as in control animals, other NCPs may compensate for some structural inadequacies of bone matrix in the osteonectin-null mutant mouse. The ONT(−/−) mouse, however, differs from mice carrying null mutations in other NCPs genes.
Normal human trabecular bone has 20- to 40-fold more ONT than cortical bone [74], which may make trabecular bone more sensitive to age-related ONT changes in the body. Senile osteoporosis in aged humans results in decreased cortical and trabecular bone. It has been proposed that polymorphism in osteonectin genes may play a role in inherited susceptibility to osteoporosis [75]. This concept appears to be supported by another mouse model, the fro/fro mouse. The fro/fro mouse displays bone fragility phenotype, and interestingly shows a lower level of osteonectin RNA and the ONT protein synthesis [76].
Fibrillins
Fibrillin-1 and -2 are ubiquitous proteins that form filamentous complexes known as microfibrils of approximately 10 nm in diameter. Fibrillins form morphologically distinct assemblies in skeletal tissues. Fibrillin microfibrils associate or interact with several other extracellular matrix proteins, including some glycoproteins and latent TGF-β-binding proteins. They crosslink elastin macromolecules with noncollagenous architectural elements of hard and soft tissue matrices [77]. A deficiency in either fibrillin-1 or -2 in mice decreases BMD through alterations of local TGF-β and bone morphogenetic proteins. Although fibrillins are not known to play a direct structural role in supporting mineral deposition in bone matrix [78], they are a part of the noncollagenous component of the bone architectural matrix [79]. These proteins are known to influence osteoblast-driven osteoclast activity [78, 80].
Recently, two types of mice mutants have been generated, one underexpressing fibrillin-1 FBN(mgR/mgR), and the other one lacking fibrillin-2 (FBN(−/−)) [81••]. Clear skeletal differences were noted between these mice as well as their respective wild-type littermates. Compared to wild-type controls, the femurs of FBN(mgR/mgR) mutant mice were longer, while those of the FBN(−/−) mutants were shorter. Furthermore, the FBN(mgR/mgR) mutants had different shape and different morphology of cortical and trabecular bone compared to the wild-type controls. Different morphology of femurs deficient or lacking fibrillin-1 or -2 demonstrated distinct modeling responses to mechanical loading with smaller effects on material quality. For example, a 4-point bending test performed on FBN(−/−) femurs demonstrated a 29% decrease in maximum load and a 30% decrease in stiffness compared to wild-type femurs. Interestingly, the FBN(mgR/mgR) femurs did not differ from wild-type animals. Thus, fibrillin-2 assemblies may have much greater impact on the mechanical properties of bone than fibrillin-1 [81••]. As mutations in fibrillin-1 and -2 cause two clinically distinct disorders (Marfan syndrome and congenital contractural arachnodactyly, respectively), an increased understanding of the role of extracellular microfibril assemblies in bone physiology can lead to the development of appropriate treatments for these diseases as well as osteoporosis.
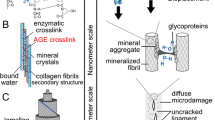
Glycation and Mechanical Integrity of Bone
Unique fracture properties of bone originate from its ability to form small-scale microdamage during load-induced absorption of energy [82–84]. The quality of bone matrix can affect the formation of particular morphologies of microdamage [82–86]. Therefore, microdamage accumulation and morphology are both risk factors for osteoporotic fractures. Various enzymatic (eg, phosphorylation, glycosylation) and nonenzymatic (eg, glycation, oxidation) post-translational modifications of extracellular bone matrix proteins and collagen are important factors of bone quality, because they influence mineralization, microdamage formation, and bone’s mechanical properties.
The collagen triplex undergoes numerous post-translational modifications and is eventually stabilized by intra- and intermolecular crosslinks formed through both enzymatic and nonenzymatic processes. For example, trivalent enzymatic crosslinks, such as pyridinoline (PYD) and deoxypyridinoline (DPD), serve as indicators of collagen maturity. They are produced during enzymatic oxidation (by lysyl oxidases) and hydroxylation (by lysyl hydrolases) of lysine residues as immature crosslinks, which subsequently undergo rearrangement to their mature forms. Hydroxylation of lysine, which is regulated by lysyl hydrolase 1, is reflected by the ratio of PYD to DPD. PYD predominate in cartilage, while DPD is more specific to bone. Normally, PYD and DPD are excreted in the urine. When bone resorption is increased, not only larger quantities of PYD and DPD are excreted in urine but the normal (∼ 4.0–8.0) ratio of PYD/DPD changes. The PYD/DPD ratio is also influenced by the patient’s health (eg, presence or absence of rheumatoid arthritis, osteoporosis, vitamin D deficiency, hyperparathyroidism, corticosteroid therapy, etc.). PYD and DPD derive mainly from bone matrix degradation and are, therefore, convenient markers of bone resorption [87••].
Biochemical, nonenzymatic changes to collagen and noncollagenous bone matrix proteins increase with aging and different bone diseases. Age-related changes in abnormal bone remodeling are primarily associated with the diminishing quality of collagen due to undesired glycation, the process that results in chemical modifications of proteins by carbohydrates [88, 89]. Advanced glycation end products (AGEs) have been shown to differ between non-fracture and fracture patients. However, the observed higher levels of AGEs in fractured bones compared to non-fracture ones do not establish causality between AGEs and fracture [10]. AGEs correlate with the stiffness of the organic matrix of bone, but it is unclear how this increased stiffness affects bone fracture toughness. The increased organic matrix stiffness can reduce microcracking and collagen deformation [90]. Consequently, changes in the matrix due to AGEs can decrease bone toughness by modifying the toughening mechanisms. More importantly, because AGEs accumulate with age, it is thought that the nonenzymatic glycation-mediated changes may help to explain age-related loss of bone toughness, differences in morphology of microdamage accumulation, and increase in bone fractures [91, 92]. We have demonstrated an increase in protein modifications through accumulation of AGEs [56••, 93] and have used AGEs to predict in vitro fracture properties measured in aged human samples [91] and in dogs treated with bisphosphonates [10].
Pentosidine (PEN) represents nonenzymatic, pentose-derived crosslink formed between lysine and arginine residues of collagen and is used as a biomarker for cumulative nonenzymatic glycation damage to proteins. Recent studies demonstrate that the measurements of PEN can predict vertebral fractures independently of BMD in vivo and in vitro [94]. However, it is noteworthy that PEN is only present in very small amounts in bone and its relationship to total fluorescent AGEs is currently unknown. Moreover, AGEs and PEN in bone account for modifications to collagen; however, the other components of the organic matrix that contribute to fracture may also undergo glycation and these have not been taken into consideration.
Conclusions
The role of noncollagenous components of the extracellular matrix in bone strength has recently begun to be recognized as an important contributor to bone fracture resistance. Current evidence indicates that together with collagen, certain major NCPs may influence bone fracture independently from bone mass. These proteins may also play a significant role in the formation of specific morphologies of microdamage. Nonenzymatic glycation is an important variable in analysis of bone’s fracture resistance, because it significantly alters bone’s organic matrix and, therefore, it may serve as a potentially useful predictor of bone fragility.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Young MF. Bone matrix proteins: their function, regulation, and relationship to osteoporosis. Osteoporos Int. 2003;14 Suppl 3:S35–42.
Schuit SC, van der Klift M, Weel AE, et al. Fracture incidence and association with bone mineral density in elderly men and women: the Rotterdam study. Bone. 2004;34:195–202.
Ott SM. When bone mass fails to predict bone failure. Calcif Tissue Int. 1993;53:S7–S13.
Sellmeyer DE. Atypical fractures as a potential complication of long-term bisphosphonate therapy. J Am Med Assoc. 2010;304:1480–3.
Yoon RS, Hwang JS, Beebe K. Long-term bisphosphonate usage and subtrochanteric insuffciency fractures: a cause for concern? J Bone Joint Surg Br. 2011;93:1289–95.
Ahlman MA, Rissing MS, Gordon L. Evolution of bisphosphonate related atypical fracture retrospectively observed with DXA scanning. J Bone Miner Res. 2012;27(2):496–8. This is a report on a patient who after 4 years of bisphosphonate treatment experienced femur fracture despite improvements in BMD..
Melton 3rd LJ, Riggs BL, Keaveny TM, et al. Structural determinants of vertebral fracture risk. J Bone Miner Res. 2007;22(12):1885–92.
Vashishth D. Rising crack growth resistance behavior in cortical bone: Implications for toughness measurements. J Biomech. 2004;37(10):943–6.
Saito M, Fujii K, Soshi S, Tanaka T. Reductions in degree of mineralization and enzymatic collagen cross-links and increases in glycation-induced pentosidine in the femoral neck cortex in cases of femoral neck fracture. Osteoporos Int. 2006;17(7):986–95.
Vashishth D. The role of the collagen matrix in skeletal fragility. Curr Osteoporos Rep. 2007;5(2):62–6.
Tang SY, Allen MR, Phipps R, et al. Changes in non-enzymatic glycation and its association with altered mechanical properties following 1-year treatment with risedronate or alendronate. Osteoporosis Int. 2009;20(6):887–94.
Sornay-Rendu E, Boutroy S, Munoz F, Delmas PD. Alterations of cortical and trabecular architecture are associated with fractures in postmenopausal women, partially independent of decreased BMD measured by DXA: the OFELY study. J Bone Miner Res. 2007;22(3):425–33.
Bornstein P. Matricellular proteins: an overview. J Cell Commun Signal. 2009;3:163–5.
Centrella M, Horowitz MC, Wozney JM, et al. Transforming growth factor-beta gene family members and bone. Endocr Rev. 1994;15:27–39.
Daughaday W, Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989;10(1):68–91.
Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16(1):3–34.
Bragdon B, Moseychuk O, Saldanha S, et al. Bone morphogenetic proteins: a critical review. Cell Signal. 2011;23:609–20. This is a comprehensive review on BMPs..
Pannicia R, Riccioni T, Zani BM, et al. calcitonin down-regulates immediate cell signals induced in human osteoclast-like cells by the bone sialoprotein-HA fragment through a post-integrin receptor mechanism. Endocrinology. 1995;136:1177–86.
Takeuchi Y, Kodama Y, Matsumoto T. Bone matrix decorin binds transforming growth factor-beta and enhances its bioactivity. J Biol Chem. 1994;269:32634–8.
Staal A, Van Wijnen AJ, Desai RK, et al. Antagonistic effects of transforming growth factor-beta on vitamin D3 enhancement of osteocalcin and osteopontin transcription: reduced interactions of vitamin D receptor/retinoid X receptor complexes with vitamin E response elements. Endocrinology. 1996;137(5):2001–11.
Keene DR, Sakai LY, Burgeson RE. Human bone contains type III collagen, type VI collagen, and fibrillin: type III collagen is present onspecific fibers that may mediate attachment of tendons, ligaments, and periosteum to calcified bone cortex. J Histochem Cytochem. 1991;39(1):59–69.
Wu JJ, Weis MA, Kim LS, et al. Differences in chain usage and cross-linking specificities of cartilage type V/XI collagen isoforms with age and tissue. J Biol Chem. 2009;284:5539–45.
McNally A, Henry Schwarcz HP, Botton GA et al.: A model for the ultrastructure of bone based on electron microscopy of ion-milled sections. PLoS One 7(1):e29258. doi:10.1371/journal.pone.0029258. In this paper a simplified model of bone mineral and collagen fibrils in fully dense cortical bone is presented.
Chen PY, Toroian D, Price PA, McKittrick J. Minerals form a continuum phase in mature cancellous bone. Calcif Tissue Int. 2011;88:351–61. In this paper the structural features of bone mineral at different length scales are described..
Singh P, Carraher C, Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol. 2010;26:397–419. doi:10.1146/annurev-cellbio-100109-104020.
Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–84. doi:10.1146/annurev.cellbio.20.010403.094555.
Durbeej M. Laminins. Cell Tissue Res. 2010;339(1):259–68. doi:10.1007/s00441-009-0838-2.
Lamoureux F, Baud’huin M, Duplomb L, et al. Proteoglycans: key partners in bone cell biology. Bioessays. 2007;29(8):758–71.
Siebers MC, ter Brugge PJ, Walboomers XF, et al. Integrins as linker proteins between osteoblasts and bone replacing materials. A critical review. Biomaterials. 2005;26(2):137–46.
LeBaron RG, Höök A, Esko JD, et al. Binding of heparin sulfate to type V collagen. J Biol Chem. 1989;264:7950–6.
Conover CA. In vitro studies of insulin-like growth factor I and bone. Growth Horm IGF Res. 2000;Supplement B:S107–10.
Conover CA. Regulation and physiological role of insulin-like growth factor binding proteins. Endocr J. 1996;43(Suppl):S43–8.
Xia Y, Schneyer AL. The biology of activin: recent advances in structure, regulation and function. J Endocrinol. 2009;202:1–12.
Aleman-Muench GR, Soldevila G. When versatility matters: activins/inhibins as key regulators of immunity. Immunol Cell Biol. 2012;90:137–48. doi:10.1038/icb.2011.32.
Jenny A, Visser JA, de Jong FH, Laven JSE, et al. Anti-Müllerian hormone: a new marker for ovarian function. Reproduction. 2006;131:1–9. doi:rep.1.00529/rep.1.00529.
Visser JA, Schipper I, Laven JSE, et al.: Anti-Müllerian hormone: an ovarian reserve marker in primary ovarian insufficiency. Nat Rev Endocrinol. 2012: doi:10.1038/nrendo.2011.224
Assoian RK, Komoriya A, Meyers CA, et al. Transforming growth factor-β in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. 1983;258:7155–60.
Seyedin SM, Thomas TC, Thompson AY, et al. Purification and characterization of two cartilage-inducing factors from bovine demineralized bone. Proc Natl Acad Sci USA. 1985;82:2267–71.
Hering S, Isken E, Knabbe C, et al. TGF-β1 and TGF-β2 mRNA and protein expression in human bone samples. Exp Clin Endocrinol Diabetes. 2001;109:217–26.
ten Dijke P, Hill CS. New insights into TGF-β-Smad signalling. Trends Biochem Sci. 2004;29:265–73.
Borton AJ, Frederick JP, Datto MB, et al. The loss of Smad3 results in a lower rate of bone formation and osteopenia through dysregulation of osteoblast differentiation and apoptosis. J Bone Miner Res. 2001;16:1754–64.
Janssens K, ten Dijke P, Janssens S, et al. Transforming growth factor-β1 to the bone. Endocr Rev. 2005;26(6):743–74.
Balooch G, Balooch M, Nalla RK, et al. TGF-β regulates the mechanical properties and composition of bone matrix. Proc Natl Acad Sci USA. 2005;102(52):18813–8.
Filvaroff E, Erlebacher A, Ye J, et al. Inhibition of TGF-beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development (Cambridge, UK). 1999;126:4267–79.
Kavukcuoglu NB, Denhardt DT, Guzelsu N, Mann AB. Osteopontin deficiency and aging on nanomechanics of mouse bone. J Biomed Mater Res. 2007;83:136–44.
Mochida Y, Parisuthiman D, Pornprasertsuk-Damrongsri S, et al. Decorin modulates collagen matrix assembly and mineralization. Matrix Biol. 2009;28:44–52.
Olszta MJ, Cheng X, Jee SS, et al. Bone structure and formation: a new perspective. Mater Sci Eng Rev. 2007;58:77–116.
Qiu SR, Wierzbicki A, Orme CA, et al. Molecular modulation of calcium oxalate crystallization by osteopontin and citrate. Proc Natl Acad Sci USA. 2004;101(7):1811–5.
Poundarik A, Karim L, Gundberg C, Vashishth, D.: The role of osteocalcin in bone fracture. Proceedings of the 55th Meeting of the Orthopaedic Research Society, Las Vegas, USA, February, 22–25, 2009 [abstract 724].
Sodek J, Ganss B, McKee MD. Osteopontin. Crit Rev Oral Biol Med. 2000;11(3):279–303.
McKee MD, Nanci A. Osteopontin deposition in remodeling bone: an osteoblast mediated event. J Bone Miner Res. 1996;11:873–4.
Rittling SR, Matsumoto HN, McKee MD, et al. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Miner Res. 1998;13(7):1101–11.
Thurner PJ, Erickson B, Jungmann R, et al. High speed photography of compressed human trabecular bone correlates whitening to microscopic damage. Eng Fract Mech. 2007;74:1928–41.
Fantner GE, Hassenkam T, Kindt JH, et al. Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture. Nat Mater. 2005;4:612–6.
Fantner GE, Adams J, Turner P, et al. Nanoscale ion mediated networks in bone: osteopontin can repeatedly dissipate large amounts of energy. Nano Lett. 2007;7:2491–8.
Sroga GE, Karim L, Colón W, Vashishth D. Biochemical characterization of major bone–matrix proteins using nanoscale-size bone samples and proteomics methodology. Mol Cell Proteomics. 2011;10(9):1–12. doi:10.1074/mcp.M110.006718. In this paper a novel strategy that permits comprehensive biochemical analysis of bone matrix proteins is presented. Application of the strategy facilitated detection of unexpected changes in the contents and composition of major bone matrix proteins, which occur during aging..
Thurner PJ, Chen CG, Ionova-Martin S, et al. Osteopontin deficiency increases bone fragility but preserves bone mass. Bone. 2010;46:1564–73. The most important result presented in this paper is a significant decrease in fracture toughness of mouse bone due to OPN deficiency..
Weinreb M, Shinar D, Rodan GA. Different pattern of alkaline phosphatase, osteopontin, and osteoealcin expression in developing rat bone visualized by in situ hybridization. J Bone Miner Res. 1990;5:831–42.
Boivin G, Morel G, Lian JB, et al. Localization of endogenous osteocalcin in neonatal rat bone and its absence in articular cartilage: effect of warfarin treatment. Virchows Arch A Pathol Anat. 1990;417:505–12.
Ducy P, Desbois C, Boyce B, et al. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382:448–52.
Kazanecki CC, Uzwiak DJ, Denhardt DT. Control of osteopontin signaling and function by post-translational phosphorylation and protein folding. J Cell Biochem. 2007;102:912–24.
Termine JD, Kleinman HK, Whitson SW, et al. Osteonectin, a bone-specific protein linking mineral to collagen. Cell. 1981;26(10, 1 Pt 1):99–105.
Delany AM, Amling M, Priemel M, et al. Osteopenia and decreased bone formation in osteonectin-defcient mice. J Clin Invest. 2000;105:915–23.
Young MF, Kerr JM, Ibaraki K, et al. Structure, expression, and regulation of the major noncollagenous matrix proteins of bone. Clin Orthop. 1992;281:275–94.
Lane TF, Sage EH. The biology of SPARC, a protein that modulated cell–matrix interactions. FASEB J. 1994;8:163–73.
Shankavaram UT, DeWitt DL, Funk SE, et al. Regulation of human monocyte matrix metalloproteinases by SPARC. J Cell Physiol. 1997;173:327–34.
Tremble PM, Lane TF, Sage EH, Werb Z. SPARC, a secreted protein associated with morphogenesis and tissue remodeling, induces expression of metalloproteinases in fibroblasts through a novel extracellular matrix-dependent pathway. J Cell Biol. 1993;121:1433–44.
Maillard C, Malaval L, Delmas PD. Immunological screening of SPARC/Osteonectin in nonmineralized tissues. Bone. 1992;13(3):257–64.
Butler W. The nature and significance of osteopontin. Connect Tiss Res. 1989;23:123–36.
Gilmour DT, Lyon GJ, Carlton MB, et al. Mice deficient for the secreted glycoprotein SPARC/osteonectin/BM40 develop normally but show severe age-onset cataract formation and disruption of the lens. EMBO J. 1998;17:1860–70.
Norose K, Clark JI, Syed NA, et al. SPARC deficiency leads to early onset cataractogenesis. Invest Ophthalmol Vis Sci. 1998;39:2674–80.
Margolis RN, Canalis E, Partridge NC. Anabolic hormones in bone: basic research and therapeutic potential. J Clin Endocrinol Metab. 1996;81:872–7.
Baron RE. Anatomy and ultrastructure of bone. In: Favus MJ, editor. Primer on the metabolic bone diseases and disorders of mineral metabolism. New York: Lippincott-Raven; 1996. p. 3–10.
Ninomiya JT, Tracy RP, Calore JD, et al. Heterogeneity of human bone. J Bone Miner Res. 1990;5:933–8.
Delany AM, McMahon DJ, Powell JS, et al. Osteonectin/SPARC polymorphisms in Caucasian men with idiopathic osteoporosis. Osteoporos Int. 2008;19(7):969–78.
Muriel MP, Bonaventure J, Stanescu R, et al. Morphological and biochemical studies of a mouse mutant (fro/fro) with bone fragility. Bone. 1991;12:241–8.
Ramirez F, Rifkin DB. Extracellular microfibrils: contextual platforms for TGF-β and BMP signaling. Curr Opin Cell Biol. 2009;21:616–22.
Nistala H, Lee-Arteaga S, Smaldone S, et al. Fibrillin-1 and -2 differentially modulate endogenous TGF-β and BMP bioavailability during bone formation. J Cell Biol. 2010;190:1107–21.
Ramirez F. Extracellular matrix in the skeleton. In: Pourquie O, editor. The skeletal system. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2009. p. 341–53.
Nistala H, Lee-Arteaga S, Smaldone S, et al. Extracellular microfibrils modulate osteoblast-supported osteoclastogenesis by restricting TGF-β stimulation of RANKL production. J Biol Chem. 2010;285:34126–33.
Arteaga-Solis E, Sui-Arteaga L, Kim M, et al. Material and mechanical properties of bones deficient for fibrillin-1 or fibrillin-2 microfibrils. Matrix Biol. 2011;30(3):188–94. In this paper, it was demonstrated that fibrillin-1 and -2 differently influence bone strength. In particular, the fibrillin-2 knockout mice bones displayed a significant decrease in the biomechanical properties..
Diab T, Condon KW, Burr DB, Vashishth D. Age-related change in the damage morphology of human cortical bone and its role in bone fragility. Bone. 2006;38(3):427–31.
Diab T, Vashishth D. Morphology, localization and accumulation of in vivo microdamage in human cortical bone. Bone. 2007;40:612–8.
Arlot M, Burt-Pichat B, Roux J-P, et al. Microarchitecture influences microdamage accumulation in human vertebral trabecualr bone. J Bone Miner Res. 2008;23(10):1613–8.
Yeni YN, Hou FJ, Ciarelli T, et al. Trabecular shear stresses predict in vivo linear microcrack density but not diffuse damage in human vertebral cancellous bone. Ann Biomed Eng. 2003;31(6):726–32.
Caler WE, Carter DR. Bone creep-fatigue damage accumulation. J Biomech. 1989;22:625–35.
Saito M, Marumo K. Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporosis Int. 2010;21:195–214. Currently, this is the most recent, comprehensive review on collagen crosslinks and their influence on bone quality..
Shiraki M, Kuroda T, Tanaka S, et al. Nonenzymatic collagen cross-links induced by glycoxidation (pentosidine) predicts vertebral fractures. J Bone Miner Metab. 2008;26(1):93–100.
Vashishth D, Gibson GJ, Khoury JI, et al. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone. 2001;28:195–201.
Vashishth D. The role of the collagen matrix in skeletal fragility. Curr Osteoporos Rep. 2007;5:62–6.
Tang SY, Zeenath U, Vashishth D. Effects of non-enzymatic glycation on cancellous bone fragility. Bone. 2007;40(4):1144–51.
Vashishth D. Hierarchy of bone microdamage at multiple length scales. Int J Fatigue. 2007;29:1024–33.
Sroga GE, Vashishth D. UPLC methodology for identification and quantitation of naturally fluorescent crosslinks in proteins: a study of bone collagen. J Chromatogr B. 2011;879:379–85. doi:10.1016/j.jchromb.2010.12.024.
Viguet-Carrin S, Roux JP, Arlot ME, et al. Contribution of the advanced glycation end product pentosidine and of maturation of type I collagen to compressive biomechanical properties of human lumbar vertebrae. Bone. 2006;39:1073–9.
Weis MA, Hudson DM, Kim L, et al. Location of 3-hydroxyproline residues in collagen types I, II, II, and V/XI implies a role in fibril supramolecualr assembly. J Biol Chem. 2010;285(4):2580–90.
Hauschka PV, Lian JB, Cole DEC, et al. Osteocalcin and matrix Gla protein: vitamin K-dependent proteins in bone. Physiol Rev. 1989;69(3):990–1047.
Hankenson KD, Bornstein P. The secreted protein thrombospondin-2 is an auticrin inhibitor of marrow stromal cell proliferation. J Bone Mineral Res. 2002;17:415–25.
George A, Veis A. Phosphorylated proteins and control over apatite nucleation, crystal growth, and inhibition. Chem Rev. 2008;108:4670–93.
Acknowledgments
The work was supported by the National Institutes of Health (NIH) grants AR4963506-11 and AG20618.
Disclosure
Conflicts of interest: G.E. Sroga: none; D. Vashishth: has received other grant support from the NIH; and has filed for a patent.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sroga, G.E., Vashishth, D. Effects of Bone Matrix Proteins on Fracture and Fragility in Osteoporosis. Curr Osteoporos Rep 10, 141–150 (2012). https://doi.org/10.1007/s11914-012-0103-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-012-0103-6