
Abstract
Purpose of the review
Bone’s ability to withstand load resisting fracture and adapting to it highly depends on the quality of its matrix and its regulators. This review focuses on the contribution of bone quality to fracture resistance and possible therapeutic targets for skeletal fragility in aging and disease.
Recent findings
The highly organized, hierarchical composite structure of bone extracellular matrix together with its (re)modeling mechanisms and microdamage dynamics determines its stiffness, strength, and toughness. Aging and disease affect the biological processes regulating bone quality, thus resulting in defective extracellular matrix and bone fragility. Targeted therapies are being developed to restore bone’s mechanical integrity. However, their current limitations include low tissue selectivity and adverse side effects.
Summary
Biological and mechanical insights into the mechanisms controlling bone quality, together with advances in drug delivery and studies in animal models, will accelerate the development and translation to clinical application of effective targeted-therapeutics for bone fragility.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Healthy bone is strong (resists inelastic deformations) and tough (resists crack propagations), and is able to adapt its architecture in response to the applied loads. Bone’s unique material properties derive from its highly organized, hierarchical composite structure. Its components, primarily collagen and hydroxyapatite, but also non-collagenous proteins (NCPs) and water, are arranged at multiple length scales, and together with bone tissue (re)modeling dynamics and microdamage mechanisms, confer bone its ability to withstand loads without deformations and fractures, and to adapt to its mechanical environment. Aging, disease, and abnormal loading conditions on bone alter its composition and disrupt its hierarchical structure, changing the mechanical environment on it and increasing bone’s vulnerability to fractures and deformities. Being able to directly target bone defects with therapies is critical for the development of effective treatments to prevent fractures in skeletal diseases and disorders. This article focuses on the contribution of bone matrix to resist fracture and highlights current and possible bone therapeutics targets.
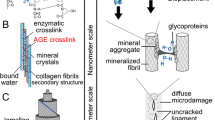
Hierarchical organization of bone and its contribution to toughness
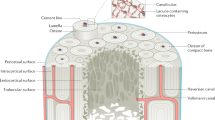
Bone dynamically adapts its shape and structure in response to the applied loads. This mechano-adaptation process is central in maintaining bone’s mechanical integrity and its ability to withstand loads, which differ with age and sex [1]. Within human bone, the matrix is organized in osteons running parallel to its longitudinal axis. Each osteon contains a central blood vessel surrounded by concentric layers of bone, the lamellae, all enclosed within thin, hypermineralized (1−5 μm) interfaces, the cement lines, generated during remodeling. Osteons orientation makes bone five times tougher to break than to split [2]. When a crack propagates, cement lines favor the formation of crack deflections and twists during bone breaking in the transverse orientation, and of crack bridging during bone splitting in the longitudinal orientation. These mechanisms increase the toughness of the bone, shielding it against the crack propagation. Cement lines further increase bone toughness through matrix slippage at their interfaces with interstitial bone [3].
Centrally in the osteons, vascular channels, known as Haversian canals, bring blood and nutrients to bone together with the Volkmann’s canals. Small mammalian bones, such those in mice, do not have osteons nor cement lines, but they have blood vessels and concentric lamellar structure around their medullar cavity. In any case, vascular porosity density within the bone matrix inversely relates to bone fracture toughness [4]. Intracortical vascular canals do not influence crack deflections, but their architecture is critical to bone resistance to fracture [5]. Many closely connected canals, as those observed, for example, in brittle osteogenesis imperfecta (OI) bone [6, 7], disrupt the continuity of bone matrix material and increase strain and stress concentrations in the bone matrix (Fig. 1A–C), thus favoring crack initiation [6] and propagation [5]. In aged human bone, the number of vascular canals, and with them of osteons and cement lines, are three times higher than in young bone. This reduces bone fracture toughness and favors the generation of microdamage accumulation in the cement lines that further facilitates bone failure [8]. Interestingly, an age-related increase in intracortical porosity has also been observed in C56BL/6 mice [9], but not in BALB/c mice, where extracellular matrix (ECM) modifications in collagen and water seem responsible for their skeletal fragility [10].

Micro- and submicron-architecture changes in OI brittle bone contribute to its fragility. A Mouse bone reconstruction with B a representative cortical bone volume finite element model loaded in compression. The yellow area shows the region of interest (ROI). C The two finite element models of representative healthy and OI cortical bone samples show the intracortical porosity of healthy and OI cortical bone. The green sites around canals and at their intersections show the location of bone within the ROI at high risk of fracture initiation when loaded at an apparent strain of 0.4%. D Second harmonic generation microscopy images of collagen fiber organization in healthy and OI cortical bone in a smaller blue ROI shows lamellar structure in healthy bone and coexistence of lamellae and woven bone in OI bone tissue. E Schematic of a bone fracture mechanics testing of a notched mouse femur. F Scanning electron microscopy of the healthy and OI bone showing their crack path during fracture. In healthy bone, the crack takes deflections, while in OI bone the crack follow a straight path, corresponding to its reduced toughening mechanisms. For more information, refer to Carriero et al. [5, 6] and Docaj et al. [18•]
At the tissue level, bone is organized in lamellae (Fig. 1D), considered as a continuous but periodically inhomogeneous layered material with variable mechanical properties depending on the orientation of their fibers [11]. Bone tissue is composed of bundles of mineralized collagen fibers arranged in a plywood-like pattern along with disorganized periods lacking this consistent distribution of fibers [12, 13•]. By virtue of its anisotropy, the interfaces between the lamellar structure may contribute to bone toughness by acting as delamination barriers, causing crack deflections (Fig. 1E, F) and twists that can double bone fracture toughness during crack growth [5, 14, 15•]. Therefore, a larger osteonal tissue with lots of lamellar area should favor fracture resistance to crack initiation and propagation [16]. In OI bone, woven bone and lamellae coexist [17], with the greater amount of woven bone in the most severe OI variants, and with OI lamellae being shorter, thinner, less organized, and smoother than in healthy bone (Fig. 1D) [17, 18•]. This altered bone structure is accompanied by reduced fracture toughness [5, 14, 18•]. Within lamellae and woven bone, osteocyte lacunae and their canaliculi network generate another level of porosity. Osteocyte density increases with high bone (re)modeling, and lacunae become more spherical in diseases [6, 19, 20], thus generating high strains in the ECM that contribute to bone fragility [21]. Similarly, strain concentrations around canaliculi promote the initiation and growth of intra-lamellar circumferential microcracks, which are associated with the formation of shear bands, and suggest an ability of bone to develop enhanced inelastic deformations by cracking control at the mineralized collagen fibril bundles level [22].
Indeed, although toughening mechanisms shielding bone from crack growth occur at its microscale, bone plasticity, which affects both strength (ductility) and initiation toughness, arises from its nanoscale. Fibrils stretching and deformation, as well as breaking of sacrificial bonds and enzymatic cross-links, allow for fibrillar sliding, intrafibrillar dilatational band formation (i.e., 100 nm long ellipsoidal voids forming between fused mineral aggregates and two adjacent NCPs), and microcracking at their interfaces [23]. Mineralized fibrils are constituted by type I collagen molecules staggered in arrays with a 67-nm offset. Aggregates of hydroxyapatite nanoplatelets form in the intrafibrillar gaps between the collagen triple helices and in extrafibrillar loci extended along the long axis of the collagen fibrils [24]. Extrafibrillar mineral inclusions in bone also exist in the form of elongated plates tenths of nanometers long and wide, and 5 nm thick, which can be either flat or curved, wrapping around collagen fibrils and forming a distinctive visual pattern resembling rosettes [13•, 24]. Most of the mineral in bone actually lie out of fibrils in the form of mineral lamellae [13•, 24]. In the presence of collagen mutations, such as in OI, decreased intrafibrillar mineralization contribute to reduced apatite crystal alignment and thinning of collagen fibrils [25•].
During elastic bone tissue deformation, the extra- and intra-fibrillar matrix stretches [26]. Damage progression and failure of the extrafibrillar matrix is responsible for initiation of bone yielding. At this point, mineralized collagen fibrils would be more involved in load bearing because the damaged extrafibrillar matrix loses its ability to carry load [26]. Mineralized collagen fibrils thus distribute the load to the nearby fibrils [27]. The load is transferred from their weaker and less mineralized overlap regions to the stronger and highly mineralized gap regions of neighboring fibrils [27].
Inside the mineralized collagen fibrils, water-mediated hydrogen bridges between hydroxyapatite and tropocollagen limit sliding between molecules and help transfer load, improving energy dissipation [28]. Loosely bound water within each fibril also contributes to bone ductility acting as a plasticizer at the interface between mineral and collagen phases in the mineralized fibril [29, 30]. When intrafibrillar water is removed, the energy dissipation of the bone is one ninth of that under hydrated conditions [30]. Structural water contained in apatite crystals has instead been suggested to boost hydroxyapatite platelets assembly and increase mineralization in bone [31•]. Hydroxyapatite significantly enhances the tensile modulus of the mineralized fibrils, reaching the highest stiffness at 2 nm thickness [32]. Thicker minerals embrittle the fibril structure [32], while fibrils with smaller mineral particles, as in type 2 diabetes mellitus (T2DM), OI and glucocorticoid-induced osteoporosis (GIOP), have a low elastic modulus [5, 33, 34•]. Increased mineral content in overlap regions likely improves bone elastic modulus, ultimate strength, and fracture toughness [27]. Fibril deformations (stretching and sliding) vary with tissue strain and highly decrease at high strain rates, with fibril deformations being primarily stretching [35]. Large yield stress triggers fibrils debonding at their interface and reduces their plastic deformation [36]. This strain-rate stiffness effect is, however, suppressed in bone with altered mineralized fibrils nanostructure, due to low shear-transfer and shear-reinforcement of short mineral plates [34•].
The quality of collagen that constitutes bone fibrils depends on its post-translational modifications, which involve covalent and enzymatic processes, and on its relationship with minerals, NCPs, and water. Hydroxylation and lysyl oxidase-mediated modifications are important processes for hydrogen bonding and cross-linking catalysis to form stable collagen molecules and fibrils, respectively. Tightly bound water, trapped inside tropocollagen molecules, participates in their conformational stability by attaching to hydroxyl groups of hydroxyproline [29, 37]. As collagen matures, interfibrillar cross-links predominate over intrafibrillar cross-links, and loosely bound water in fibrils is substituted by mineral. Thus, water content in bone decreases with age [10, 38•], reducing its plasticity [30, 37, 39]. Inhibition of lysyl oxidase results in cross-link perturbations and decreased fracture toughness [40]. Enzymatic cross-linking varies throughout the skeleton as a function of turnover rate and mechanical environment on bone [41]. Mechanical loading on bone helps in stabilizing the collagen matrix through divalent cross-links maturation [41]. In diabetic bone, hyperglycemia impairs the formation of divalent cross-links because collagen has increased maturity despite a reduced amount of enzymatic cross-links [42]. A type of enzymatic hydroxylysine glycosylation, namely glucosylgalactosylhydroxylysine (GGHL), is overexpressed in OI collagen as a sign of retarded triple helix formation [43] and may hinder divalent enzymatic cross-links maturation [44].
Non-enzymatic glycation (NEG) of collagen, involving the attachment of a free sugar to a collagen amino group, disrupts bone matrix quality and reduces its crack growth toughness by 35% [45]. Glycation widens intermolecular space, hampering fibril packing, and depresses lysyl oxidase-driven cross-linking as glucose carbonyls (advanced glycation end-products (AGEs) precursors) and telopeptide aldehydes (enzymatic cross-link precursors) target the same triple-helical sites [46•]. Glycated organic matrix has a lower capacity to sustain deformation and exhibits a lower creep; therefore, the ability of bone to dissipate energy is reduced and it generates longer microcracks that coalesce, resulting in longer crack length [45]. In aging and disease, bone fibril over-cross-linking by AGEs formation due to glycation, oxidative stress, or low bone turnover inversely correlates with fracture toughness [5, 8, 47]. In most cases, high AGEs content has been associated with increased resistance to fibril deformation, reduced fibrillar sliding [48], and compromised bone plasticity and resistance to fractures [8]. However, in OI, the role of AGEs remains unclear as increased deformations of the mineralized fibrils are observed in weak mineralized fibrils and brittle bone [5]. AGEs breakers, alagebrium [49•] and n-acetylcysteine [50•], did not improve bone mechanics, although they reduced bone AGEs content and oxidative stresses in rats with chronic kidney disease (CKD). Aminoguanidine and pyridoxamine tested in glucose treated cadaveric bone prevented AGEs formation and subsequent biomechanical bone degradation [51]. These studies assessed NEG as a measurement of pentosidine or fluorescent AGEs concentration. However, the amount of pentosidine is very small in bone, and carboxymethyl-lysine (CML) and glucosepane, which are not fluorescent, are found in higher concentrations (>40 times more) [52•]. Accumulation of CML in human bone strongly inversely correlates with crack propagation toughness [52•]. Further investigation is therefore needed to fully understand the impact of all NEGs on bone fracture resistance to crack initiation and growth. These studies may also lead to new diagnostic assays and therapeutic approaches for bone fragility. An age-dependent reduction in fracture resistance of cortical bone has been observed in the presence of deamidation of asparagine and glutamine residues, which disturb the affinity of collagen triple helices to bind with each other in favor of water bonds, increasing fibril diameter, and reducing bone fracture toughness [53•]. More studies are needed to better understand the contribution of deamidation to bone fragility, and how it varies in disease. This may open new avenues for targeted therapies for bone fragility.
Molecular interactions between mineralized collagen fibrils and extra-fibrillar mineral aggregates involving NCPs play a critical role in bone quality. Particularly, osteopontin (OPN) and osteocalcin (OC) at the interfibrillar interface constitute less than 2–3% of bone weight but contribute for more than 30% to its fracture toughness by a synergistic deformation mechanism of the two NCPs and bone [23, 54•]. Through strong anchoring and formation of dynamic binding sites on mineral nanoplatelets, bone nanointerface can achieve large non-linear deformation and great ductility [54•]. Whereas the rigid OC binds tightly to hydroxyapatite crystals and remains mostly stationary during deformation due to its high affinity to the mineral surface, OPN participates in the cleavage and reformation of the sacrificial bonds, detaching from one OC and rolling towards neighboring OCs in the shear direction [23, 54•, 55]. Additionally, OPN can generate new binding regions with the hydroxyapatite surface [54•]. The further stretching and unraveling leading to OPN denaturation entails dilatational bands formation, dissipating large amounts of energy [23, 54•, 56]. The one-third reduction in bone toughness in OPN and OC knockout mice has been associated with the absence of dilatational bands and subsequent diffuse damage formation, along with increased calcium variability [23, 57]. OC function can be affected by collagen glycation, forfeiting much of its energy dissipation ability [55]. Similar compromised bone toughness is experienced with loss of proteoglycans (PGs) and glycosaminoglycans, coupled with a decrease of bound water, such as in aging [38•, 58]. OPN phosphorylation adds additional negative charge and hydrophilicity that potentially eases its adhesion to mineral [59•], while its binding to free calcium ions may prevent unwanted adsorption to other components and facilitate its deposition on mineral surface [60]. The absence of NCPs triggers alterations in mineralization degree, fibrillogenesis, bone formation, and microdamage accumulation [58, 61, 62]. For example, deletion of decorin and/or biglycan results in highly disorganized collagen fibrils [63]. Thus, by targeting NCPs, or key regulatory pathways controlling their expression and activity, bone quality can be affected directly by altering its fracture toughness or indirectly by regulating mineralized collagen fibrils organization or bone cellular activity. For instance, administration of chondroitin sulfate (CS) has been shown to have antioxidant, anticatabolic, hypoglycemic, and antidiabetic effects [64], and to improve bound water amount and bone toughness [58].
Table 1 summarizes alterations in collagen and mineral structure and composition due to aging and diseases, which affect bone hierarchical structure and mechanical competences to sustain loads and resist fractures.
The regulators of bone matrix quality and targeted therapies for bone fragility
Mechanical loading on bone increases microdamage concentration within its matrix in the form of linear and diffuse microcracks. Linear microcracks, a micron-scale damage, boost bone remodeling by inducing osteocyte apoptosis possibly by directly cutting the osteocyte processes, or indirectly by changing the lacunar–canalicular fluid flow in the damaged area [90, 91]. Osteocyte apoptosis triggers surrounding surviving osteocytes to produce RANKL, a cytokine promoting osteoclastic bone resorption by binding to RANK and regulating osteoclasts fission into osteomorphs, bone cells able to merge back into new osteoclasts with little energy expenditure as needed during bone remodeling [92••]. In addition, hydrogen ions and cathepsin K aid in bone demineralization and collagen degradation, respectively. Mesenchymal stem cells (MSCs) are attracted to the repair site by osteoclast-secreted transforming growth factor-β (TGF-β), and osteocyte-produced insulin-like growth factor-1 (IGF-1) triggers their differentiation into osteoblasts [93••]. At the mineral level, the tissue-nonspecific alkaline phosphatase (TNAP) enzyme and the progressive ankylosis protein (ANK) regulate extracellular matrix mineralization by controlling levels of osteocyte- and osteoblast-secreted inorganic pyrophosphate (PPi), a potent mineralization inhibitor [94]. The loss of TNAP and ANK function can cause hypo- and hyper-mineralization, respectively [94, 95].
Aging and disease alter bone remodeling (Table 1). Inhibition of bone remodeling affects its extracellular matrix quality and resistance to fracture: microcracks accumulate within their tissues, and mineral, collagen maturity, and AGEs content within the fibrils increase, thus reducing fibrillar sliding and bone plasticity [8, 71]. Age- and disease-related declines in the number of osteocytes and canaliculi may reduce their mechanosensory ability to detect microdamage and the release of RANKL to initiate repair, leading to the accumulation of microcracks [96]. On the other hand, increased bone remodeling may affect bone mineralization and produce a matrix with osteoid layers next to heavily mineralized regions, as in vitamin-D deficiency [83]. Osteoclasts cannot go through thick osteoid layers, and the bone underneath continues to age and mineralize although overall bone mineral content progressively decreases [83]. In OI bone, tissue hypermineralization with reduced carbonate content (as a sign of crystals immaturity) may favor the formation of microcracks, further triggering bone turnover [5] that contributes to bone fragility.
Diffuse damage, made of submicron-sized cracks, does not cause osteocyte apoptosis nor trigger osteocytic or osteoclastic activity, but self-repairs [97] spontaneously through a physicochemical remineralization process [98••], similar to that occurring in enamel repair. It is possible that the lacunar–canalicular system of osteocytes plays a role in the maintenance of bone tissue ionic fluid and in the transport of minerals and other adhesive proteins across the matrix to sites of damage, required for the chemical repair of diffuse damage [98••]. Further studies are required to understand the actual mechanisms of this self-healing and if aging and disease alter this process, and its contribution to bone fracture risk.
Osteocytic osteolysis or perilacunar remodeling (PLR) has been suggested to have a role in determining and maintaining bone quality [99]. This process has the potential to not only regulate mineral homeostasis but also to release calcium from the matrix, affect osteocyte mechanosensation, and alter bone remodeling. Osteocyte-mediated proton release demineralizes the perilacunar matrix and frees calcium ions, and in turn metalloproteinase-13 (MMP-13), tartrate resistance acid phosphate (TRAP), and cathepsin K remove the organic phase. Disruption of osteocyte-mediated resorption causes bone fragility in MMP-13 knock-out mice, owing to a highly porous, small cortex with disorganized canaliculi, increased collagen cross-linking, and smaller crystals with low crystallinity [95]. Compromised bone quality is also present in mice with knock-out and overexpressed tissue inhibitor of metalloproteinase-3 (TIMP-3) [100, 101], a MMP-13 inhibitor [100]. Particularly, TIMP-3 deficiency increases cortical porosity, acid phosphate levels, and heterogeneity of the collagen cross-link profile, and decreased carbonate-to-phosphate ratio [101]. Interestingly, PLR happening during lactation shows a recoverable reduction in bone tissue-level elastic modulus that has been attributed to changes in lacunar and canalicular space [102]. The extent and mechanisms of bone repair due to PLR remain to be revealed.
Microcracks, possibly due to masticatory stresses, also occur in ear ossicles and the otic capsule, where they accumulate over time due to downregulated bone remodeling and PLR. It has been suggested that mechanisms controlling bone remodeling and PLR in the otic capsule are different from those in the long bones, perhaps to preserve auditory function [103]. Increased porosity of the ear bone, as occurs in OI and otosclerosis, can further increase the formation of microcracks and bone fractures in the ears [104•]. The presence of zinc at sites of ossification in the ear seems to help prevent cochlear damage, while its deficiency in aging mice potentiate hearing loss [105]. Further studies are needed to elucidate the functional implications of biological controllers for bone matrix quality and their relationship with genetic expression at different body sites and in bone diseases with secondary disabilities, such as hearing loss.
The combination of bone remodeling, PLR, and diffuse damage healing plays an essential role in maintaining bone compositional, structural, and mechanical integrity. The interconnection between bone cells and matrix is regulated by intra- and extra-cellular molecular-signaling pathways that ultimately control bone quality. When these are disrupted, bone becomes fragile. Proteins and signaling pathways may either directly affect the mineral or organic constituents of bone ECM or have an effect on the cellular activity that regulates bone repair and (re)modeling, impairing bone mechanical properties. Endocrine, paracrine, and autocrine pathways are responsible for the cellular response to signaling in the processes regulating bone quality. This signaling may activate multiple cell populations independently or in a cascade of events with mutual cell interactions. Several growth factors regulating bone repair are likewise involved in determining bone quality. For example, TGF-β is essential for maintaining bone quality by coupling the activity of osteoblasts and osteoclasts, and human mutations in many TGF-β pathway components have been associated with skeletal dysplasia and disease [106]. TGF-β couples with other factors playing pivotal roles in bone homeostasis and cell cycle regulation, and their regulation offers positive results in terms of correcting aberrant bone remodeling. Table 2 reports the major bone proteins, signaling pathways, and transcription regulatory networks known to be implicated in controlling bone ECM material properties, offering targets for bone therapies.
Despite the advances in the field, there is a critical need to fully elucidate the effect of bone cells and biology on controlling bone quality, and particularly its fragility. This will suggest new targets for the development of therapies to prevent bone fragility. In this regard, the combination of genetically modified rodents with investigations of bone fracture toughness offers the opportunity to further understand the molecular control of bone material properties and quality. Our research and that of others on bone fracture toughness has disclosed mechanisms of bone fragility in OI mice, CKD rats, and diabetic ZDSD rats [5, 6, 14, 47, 53•, 89, 104•, 166], as well as the importance of OPN, OC, PHOSPHO1, and TIMP-3 for bone quality and toughness [23, 57, 100, 101, 162, 163]. However further research is needed in this direction, and studies must help unveil the complex relationship between bone biology and mechanics. Studies on cell activity and their signaling can shed a light on their influence on bone quality. For example, increased bone fragility in OI has recently not only related to its extracellular matrix impairment but also to altered intracellular homeostasis due to mutant collagen retention [152, 153]. Endoplasmic reticulum stress modulates the OI phenotype severity in its Brtl mouse model, and activates the unfolded protein response, autophagy, and apoptosis in human fibroblasts in dominant forms of OI as well as in some recessive OI forms characterized by altered collagen synthesis [152, 153]. Treatment with 4-phenylbutyrate (4PBA) chemical chaperone ameliorates OI cells homeostasis in vitro, and improves the OI bone phenotype in the Chihuahua zebrafish OI model by reducing intracellular misfolded protein accumulation and promoting protein secretion [152, 153]. However, more research is needed to investigate the material properties of the 4PBA-treated OI bone and enhance therapy efficacy by effectively deliver the drug to bone. Similarly, further studies on the recently discovered osteomorph genes, controlling bone structure and function, with their upregulation being associated with human skeletal diseases and osteopenia [92••], may further unveil new possibilities for bone fragility treatment. Also, bone has recently been revealed to play an important role in regulating glucose metabolism through the release of osteokines [167], with increased plasma OPN and OPG levels in prediabetes pathogenesis. While the role of OC in regulating glucose metabolism is unknown [167], its levels inversely correlated with circulating free fatty acids concentration [88•]. Future research may consider the development of novel therapies targeting these biomarkers for aging, T2DM, and osteoporosis populations. Table 2 presents existing and possible targeted therapeutic treatments for the major known regulators of bone quality, and their effect on bone remodeling and matrix properties.
Nowadays, bone treatments are essentially still systemic, requiring mainly oral administration or injections. Their main challenges are low bone tissue selectivity, with high doses taken for the drug to reach bone, and safety, due to their adverse side effects [168]. Moreover, once the treatment reaches bone, its low permeability and reduced blood flow may further hinder the drug efficacy. Treatments for bone fragility have so far mostly relied on bisphosphonates, a group of antiresorptive drugs, that can induce atypical femur fractures by long-term low bone turnover and/or jaw osteonecrosis, as well as gastrointestinal adverse effects and cancer of the esophagus from oral treatments, and/or atrial fibrillation due to increased blood calcium level [93••]. Similarly, some bone anabolic drugs stimulate its formation by binding to the parathyroid hormone type I receptor, but they can cause post-dose hypercalcemia and increase risk of developing osteosarcoma [169]. Therefore, there is a critical need to improve drug delivery at the appropriate concentrations directly to bone. Targeted therapy delivered to either bone matrix components or cell signaling, and activation can improve efficacy of treatments, while reducing dosage and systemic toxicity-related side effects. Nanosystems, such as alendronate conjugated nanodiamonds (ALN-NDs), extend clinical exposure while reducing side effects, whereas selective estrogen receptor modulators (SERMs), such as Raloxifene, have estrogen-like resorptive actions in bone, but neutral effects in other tissues, e.g., breast and uterus, overcoming the problem of low tissue selectivity for estrogens. As opposed to antiresorptive treatment, anabolic therapy enhances bone formation rather than reducing bone resorption. When combined with the collagen-binding domain (CBD), parathyroid hormone (PTH) treatment exerts a long-lasting bone anabolic effect while preventing inconvenient undesirable effects (e.g., hypercalcemia) [93••].
New forms of drug delivery for bone-targeted therapies have been tested in laboratories that promise to improve therapeutic efficacy, control drug release, and reduce systemic toxicity. For instance, cell-infiltratable and injectable gelatin hydrogels encapsulating MSCs successfully fostered bone regeneration in an animal model of steroid-associated osteonecrosis [170], while polyurethane nanomicelles can embody and release miRNAs to osteoclasts at the bone resorption surface, improving bone microarchitecture in ovariectomized osteoporotic mice [171]. However, most non-responsive nanocarriers cannot accomplish a realistic delivery of their payload to the target site. In such a case, enzyme-responsive delivery systems exploit altered expression of specific enzymes, such as cathepsin K and certain MMPs, to drive the liberation of drugs [172].
Conclusions
With a comprehensive understanding of the biological mechanisms controlling bone quality, particularly toughness, and the development of new biotechnology for drug delivery, novel bone-targeted therapies for bone fragility will improve in the future, holding potential for their use in the clinic. A greater understanding of the physiological differences between humans and animals affecting bone mechanics, as well as advance in bone cell biology, genetics, and genomics will accelerate the translation of bone targeted-therapy to clinical application.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Carriero A, Javaheri B, Bassir Kazeruni N, Pitsillides A, Shefelbine S. Age and sex differences in load-induced tibial cortical bone surface strain maps: JBMR Plus; 2021. This article quantifies how the mechanical environment on bone varies with age and sex in C57BL6 mice.
• Ritchie RO. How does human bone resist fracture? Ann N Y Acad Sci. 2010;1192:72.
Skedros JG, Keenan KE, Williams TJ, Kiser CJ. Secondary osteon size and collagen/lamellar organization ("osteon morphotypes") are not coupled, but potentially adapt independently for local strain mode or magnitude. J Struct Biol. 2013;181(2):95–107.
Yeni YN, Brown CU, Wang Z, Norman TL. The influence of bone morphology on fracture toughness of the human femur and tibia. Bone. 1997;21(5):453–9.
Carriero A, Zimmermann EA, Paluszny A, Tang SY, Bale H, Busse B, et al. How tough is brittle bone? Investigating osteogenesis imperfecta in mouse bone. J Bone Miner Res. 2014;29(6):1392–401.
Carriero A, Doube M, Vogt M, Busse B, Zustin J, Levchuk A, et al. Altered lacunar and vascular porosity in osteogenesis imperfecta mouse bone as revealed by synchrotron tomography contributes to bone fragility. Bone. 2014;61:116–24.
Jameson JR, Albert CI, Busse B, Smith PA, Harris GF. 3D micron-scale imaging of the cortical bone canal network in human osteogenesis imperfecta (OI). Medical imaging 2013: Biomedical applications in molecular, structural, and functional imaging; 2013: International Society for Optics and Photonics.
Zimmermann EA, Busse B, Ritchie RO. The fracture mechanics of human bone: influence of disease and treatment. BoneKEy Rep. 2015;4.
Ferguson VL, Ayers RA, Bateman TA, Simske SJ. Bone development and age-related bone loss in male C57BL/6J mice. Bone. 2003;33(3):387–98.
Creecy A, Uppuganti S, Girard MR, Schlunk SG, Amah C, Granke M, et al. The age-related decrease in material properties of BALB/c mouse long bones involves alterations to the extracellular matrix. Bone. 2020;130:115126.
Razi H, Predan J, Fischer FD, Kolednik O, Fratzl P. Damage tolerance of lamellar bone. Bone. 2020;130:115102.
Reznikov N, Shahar R, Weiner S. Three-dimensional structure of human lamellar bone: The presence of two different materials and new insights into the hierarchical organization. Bone. 2014;59:93–104.
• Grandfield K, Vuong V, Schwarcz HP. Ultrastructure of bone: hierarchical features from nanometer to micrometer scale revealed in focused ion beam sections in the TEM. Calcif Tissue Int. 2018;103(6):606–16. This article demonstrates the presence of curved mineral lamellae and presents a better insight into how bone is organized at the nanometer scale.
Carriero A, Zimmermann EA, Shefelbine SJ, Ritchie RO. A methodology for the investigation of toughness and crack propagation in mouse bone. J Mech Behav Biomed Mater. 2014;39:38–47.
• Docaj A, Jeong MS, Zimmermann EA, Ritchie RO, Carriero A. Age effect on bone toughness in osteogenesis imperfecta. Trans Ann Meet Orthopaedic Res Soc; 2020. This conference proceeding abstract presents changes in bone fracture toughness during skeletal growth in healthy and osteogenesis imperfecta mice.
Granke M, Makowski AJ, Uppuganti S, Nyman JS. Prevalent role of porosity and osteonal area over mineralization heterogeneity in the fracture toughness of human cortical bone. J Biomech. 2016;49(13):2748–55.
Shapiro F, Maguire K, Swami S, Zhu H, Flynn E, Wang J, et al. Histopathology of osteogenesis imperfecta bone. Supramolecular assessment of cells and matrices in the context of woven and lamellar bone formation using light, polarization and ultrastructural microscopy. Bone Rep. 2021;14:100734.
Docaj A, Carriero A. Site-specific changes in collagen orientation in osteogenesis imperfecta mouse bone. Eur Soc Biomechan. 2021.
Heveran CM, Schurman CA, Acevedo C, Livingston EW, Howe D, Schaible EG, et al. Chronic kidney disease and aging differentially diminish bone material and microarchitecture in C57Bl/6 mice. Bone. 2019;127:91–103.
van Hove RP, Nolte PA, Vatsa A, Semeins CM, Salmon PL, Smit TH, et al. Osteocyte morphology in human tibiae of different bone pathologies with different bone mineral density — Is there a role for mechanosensing? Bone. 2009;45(2):321–9.
De Paolis A, Jeong S, Cardoso L, Carriero A. Lacuna morphology affects strains in the cell body and dendrites, and on the bone tissue. Eur Soc Biomechan. 2019.
Ebacher V, Guy P, Oxland TR, Wang R. Sub-lamellar microcracking and roles of canaliculi in human cortical bone. Acta Biomater. 2012;8(3):1093–100.
Poundarik AA, Diab T, Sroga GE, Ural A, Boskey AL, Gundberg CM, et al. Dilatational band formation in bone. Proc Natl Acad Sci U S A. 2012;109(47):19178–83.
Schwarcz H, Binkley D, Luo L, Grandfield K. A search for apatite crystals in the gap zone of collagen fibrils in bone using dark-field illumination. Bone. 2020;135:115304.
Maghsoudi-Ganjeh M, Samuel J, Ahsan AS, Wang X, Zeng X. Intrafibrillar mineralization deficiency and osteogenesis imperfecta mouse bone fragility. J Mechan Behav Biomed Mater. 2021:104377. This article demonstrates a decrease in intrafibrillar mineralization in the osteogenesis imperfecta murine model, and suggest this as one of the causes of its reduced toughness.
Lin L, Samuel J, Zeng X, Wang X. Contribution of extrafibrillar matrix to the mechanical behavior of bone using a novel cohesive finite element model. J Mech Behav Biomed Mater. 2017;65:224–35.
Wang Y, Ural A. A finite element study evaluating the influence of mineralization distribution and content on the tensile mechanical response of mineralized collagen fibril networks. J Mech Behav Biomed Mater. 2019;100:103361.
Nair AK, Gautieri A, Buehler MJ. Role of intrafibrillar collagen mineralization in defining the compressive properties of nascent bone. Biomacromolecules. 2014;15(7):2494–500.
Granke M, Does MD, Nyman JS. The role of water compartments in the material properties of cortical bone. Calcif Tissue Int. 2015;97(3):292–307.
Maghsoudi-Ganjeh M, Wang X, Zeng X. Computational investigation of the effect of water on the nanomechanical behavior of bone. J Mech Behav Biomed Mater. 2020;101:103454.
• Von Euw S, Chan-Chang T-H-C, Paquis C, Haye B, Pehau-Arnaudet G, Babonneau F, et al. Organization of bone mineral: the role of mineral-water interactions. Geosciences. 2018;8:466. This article demonstrates that structural water contained in apatite serve the mineral particles to organize themselves and increase mineralization.
Qin Z, Gautieri A, Nair AK, Inbar H, Buehler MJ. Thickness of hydroxyapatite nanocrystal controls mechanical properties of the collagen–hydroxyapatite interface. Langmuir. 2012;28(4):1982–92.
Shahar R, Weiner S. Open questions on the 3D structures of collagen containing vertebrate mineralized tissues: A perspective. J Struct Biol. 2018;201(3):187–98.
• Xi L, De Falco P, Barbieri E, Karunaratne A, Bentley L, Esapa C, et al. Bone matrix development in steroid-induced osteoporosis is associated with a consistently reduced fibrillar stiffness linked to altered bone mineral quality. Acta Biomater. 2018;76:295–307. This article demonstrates that mineralized fibrils exhibit strain-rate dependent stiffness in healthy mouse bones but not in steroid-induced osteoporotic bone having small mineral platelets.
Zimmermann EA, Gludovatz B, Schaible E, Busse B, Ritchie RO. Fracture resistance of human cortical bone across multiple length-scales at physiological strain rates. Biomaterials. 2014;35(21):5472–81.
Xu M, An B. An analysis of fracture in staggered mineralized collagen fibril arrays. Int J Solids Struct. 2020;193–194:535–49.
Unal M, Akkus O. Raman spectral classification of mineral- and collagen-bound water's associations to elastic and post-yield mechanical properties of cortical bone. Bone. 2015;81:315–26.
• Wang X, Hua R, Ahsan A, Ni Q, Huang Y, Gu S, et al. Age-related deterioration of bone toughness is related to diminishing amount of matrix glycosaminoglycans (Gags). JBMR Plus. 2018;2(3):164–73. This article demonstrates that loss of glycosaminoglycans and the subsequent loss of bound water are one of the origins of age-related deterioration of bone quality.
Fielder M, Nair AK. Effects of hydration and mineralization on the deformation mechanisms of collagen fibrils in bone at the nanoscale. Biomech Model Mechanobiol. 2019;18(1):57–68.
McNerny EM, Gong B, Morris MD, Kohn DH. Bone fracture toughness and strength correlate with collagen cross-link maturity in a dose-controlled lathyrism mouse model. J Bone Miner Res. 2015;30(3):455–64.
Gauthier R, Follet H, Langer M, Gineyts E, Rongiéras F, Peyrin F, et al. Relationships between human cortical bone toughness and collagen cross-links on paired anatomical locations. Bone. 2018;112:202–11.
Hunt HB, Pearl JC, Diaz DR, King KB, Donnelly E. Bone tissue collagen maturity and mineral content increase with sustained hyperglycemia in the KK-Ay murine model of type 2 diabetes. J Bone Miner Res. 2018;33(5):921–9.
Taga Y, Kusubata M, Ogawa-Goto K, Hattori S. Site-specific quantitative analysis of overglycosylation of collagen in osteogenesis imperfecta using hydrazide chemistry and SILAC. J Proteome Res. 2013;12(5):2225–32.
Terajima M, Perdivara I, Sricholpech M, Deguchi Y, Pleshko N, Tomer KB, et al. Glycosylation and cross-linking in bone type i collagen. J Biol Chem. 2014;289(33):22636–47.
Poundarik AA, Wu PC, Evis Z, Sroga GE, Ural A, Rubin M, et al. A direct role of collagen glycation in bone fracture. J Mech Behav Biomed Mater. 2015;52:120–30.
• Hudson DM, Archer M, King KB, Eyre DR. Glycation of type I collagen selectively targets the same helical domain lysine sites as lysyl oxidase–mediated cross-linking. J Biol Chem. 2018;293(40):15620–7. This article demonstrates that glycation products and enzymatic cross-link precursors target the same amino acids in the triple helical region of tropocollagen molecules.
Allen MR, Newman CL, Chen N, Granke M, Nyman JS, Moe SM. Changes in skeletal collagen cross-links and matrix hydration in high- and low-turnover chronic kidney disease. Osteoporos Int. 2015;26(3):977–85.
Gautieri A, Passini FS, Silván U, Guizar-Sicairos M, Carimati G, Volpi P, et al. Advanced glycation end-products: Mechanics of aged collagen from molecule to tissue. Matrix Biol. 2017;59:95–108.
• Chen NX, Srinivasan S, O'Neill K, Nickolas TL, Wallace JM, Allen MR, et al. Effect of Advanced Glycation End-Products (AGE) Lowering drug ALT-711 on biochemical, vascular, and bone parameters in a rat model of CKD-MBD. J Bone Miner Res. 2020;35(3):608–17. This article demonstrates that alagebrium administration decreases total AGE cross-link levels, yet the mechanical properties are not normalized in the Cy/+ rat model of chronic kidney disease.
• Allen MR, Wallace J, McNerney E, Nyman J, Avin K, Chen N, et al. N-acetylcysteine (NAC), an anti-oxidant, does not improve bone mechanical properties in a rat model of progressive chronic kidney disease-mineral bone disorder. PLoS One. 2020;15(3):e0230379 This article demonstrates that N-acetylcysteine administration reduces TBARS oxidative stress markers and AGE cross-link levels in the Cy/+ rat model of chronic kidney disease.
Abar O, Dharmar S, Tang SY. The effect of aminoguanidine (AG) and pyridoxamine (PM) on ageing human cortical bone. Bone Joint Res. 2018;7(1):105–10.
Thomas CJ, Cleland TP, Sroga GE, Vashishth D. Accumulation of carboxymethyl-lysine (CML) in human cortical bone. Bone. 2018;110:128–33. This article demonstrates that carboxymethyl-lysine is a non-cross-linking AGE that is abundant in bone and impairs bone mechanical properties.
• Creecy A, Brown KL, Rose KL, Voziyan P, Nyman JS. Post-translational modifications in collagen type I of bone in a mouse model of aging. Bone. 2021;143:115763. This article demonstrates how aging affects post-translational modifications in bone, with particular emphasis on the deamidation of specific residues, and how this relates to bone fracture toughness.
• Morsali R, Dai Z, Wang Y, Qian D, Minary-Jolandan M. Deformation mechanisms of "two-part" natural adhesive in bone interfibrillar nano-interfaces. ACS Biomater Sci Eng. 2019;5(11):5916–24. This article presents new insights into the deformation mechanism between OC and OPN at the interfibrillar interface in bone.
Tavakol M, Vaughan TJ. The structural role of osteocalcin in bone biomechanics and its alteration in type-2 diabetes. Sci Rep. 2020;10(1):17321.
Wang Z, Vashishth D, Picu RC. Bone toughening through stress-induced non-collagenous protein denaturation. Biomech Model Mechanobiol. 2018;17(4):1093–106.
Thurner PJ, Chen CG, Ionova-Martin S, Sun L, Harman A, Porter A, et al. Osteopontin deficiency increases bone fragility but preserves bone mass. Bone. 2010;46(6):1564–73.
Hua R, Ni Q, Eliason TD, Han Y, Gu S, Nicolella DP, et al. Biglycan and chondroitin sulfate play pivotal roles in bone toughness via retaining bound water in bone mineral matrix. Matrix Biol. 2020;94:95–109.
• Sroga GE, Vashishth D. Phosphorylation of extracellular bone matrix proteins and its contribution to bone fragility. J Bone Miner Res. 2018;33(12):2214–29. This article demonstrates that the total phosphorylation of bone matrix proteins decreases with age, with a significant impact on fracture toughness.
Kläning E, Christensen B, Sørensen ES, Vorup-Jensen T, Jensen JK. Osteopontin binds multiple calcium ions with high affinity and independently of phosphorylation status. Bone. 2014;66:90–5.
Morgan S, Poundarik AA, Vashishth D. Do Non-collagenous Proteins Affect Skeletal Mechanical Properties? Calcif Tissue Int. 2015;97(3):281–91.
Depalle B, McGilvery CM, Nobakhti S, Aldegaither N, Shefelbine SJ, Porter AE. Osteopontin regulates type I collagen fibril formation in bone tissue. Acta Biomater. 2021;120:194–202.
Kram V, Shainer R, Jani P, Meester JAN, Loeys B, Young MF. Biglycan in the Skeleton. J Histochem Cytochem. 2020;68(11):747–62.
Zheng HX, Chen J, Zu YX, Wang EZ, Qi SS. Chondroitin sulfate prevents STZ Induced diabetic osteoporosis through decreasing blood glucose, antioxidative stress, anti-inflammation and OPG/RANKL expression regulation. Int J Mol Sci. 2020;21(15).
Boskey AL, Imbert L. Bone quality changes associated with aging and disease: a review. Ann N Y Acad Sci. 2017;1410(1):93–106.
Busse B, Djonic D, Milovanovic P, Hahn M, Püschel K, Ritchie RO, et al. Decrease in the osteocyte lacunar density accompanied by hypermineralized lacunar occlusion reveals failure and delay of remodeling in aged human bone. Aging Cell. 2010;9(6):1065–75.
Adele B. Bone mineral crystal size. Osteoporos Int. 2003;14(5):16–21.
Burr DB. Changes in bone matrix properties with aging. Bone. 2019;120:85–93.
Aido M, Kerschnitzki M, Hoerth R, Checa S, Spevak L, Boskey AL, et al. Effect of in vivo loading on bone composition varies with animal age. Exp Gerontol. 2015;63:48–58.
McCreadie BR, Morris MD, Chen T-C, Sudhaker Rao D, Finney WF, Widjaja E, et al. Bone tissue compositional differences in women with and without osteoporotic fracture. Bone. 2006;39(6):1190–5.
Saito M, Marumo K. Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos Int. 2010;21(2):195–214.
Zimmermann EA, Schaible E, Gludovatz B, Schmidt FN, Riedel C, Krause M, et al. Intrinsic mechanical behavior of femoral cortical bone in young, osteoporotic and bisphosphonate-treated individuals in low- and high energy fracture conditions. Sci Rep. 2016;6(1):21072.
Brennan O, Kuliwaba JS, Lee TC, Parkinson IH, Fazzalari NL, McNamara LM, et al. Temporal changes in bone composition, architecture, and strength following estrogen deficiency in osteoporosis. Calcif Tissue Int. 2012;91(6):440–9.
Turunen MJ, Kaspersen JD, Olsson U, Guizar-Sicairos M, Bech M, Schaff F, et al. Bone mineral crystal size and organization vary across mature rat bone cortex. J Struct Biol. 2016;195(3):337–44.
Comelekoglu U, Bagis S, Yalin S, Ogenler O, Yildiz A, Sahin NO, et al. Biomechanical evaluation in osteoporosis: ovariectomized rat model. Clin Rheumatol. 2007;26(3):380–4.
Mathavan N, Turunen MJ, Tägil M, Isaksson H. Characterising bone material composition and structure in the ovariectomized (OVX) rat model of osteoporosis. Calcif Tissue Int. 2015;97(2):134–44.
Peng Z, Tuukkanen J, Zhang H, Jämsä T, Väänänen H. The mechanical strength of bone in different rat models of experimental osteoporosis. Bone. 1994;15(5):523–32.
Morello R. Osteogenesis imperfecta and therapeutics. Matrix Biol. 2018;71:294–312.
Imbert L, Aurégan J-C, Pernelle K, Hoc T. Microstructure and compressive mechanical properties of cortical bone in children with osteogenesis imperfecta treated with bisphosphonates compared with healthy children. J MechanBehav Biomed Mater. 2015;46:261–70.
Vardakastani V, Saletti D, Skalli W, Marry P, Allain J-M, Adam C. Increased intra-cortical porosity reduces bone stiffness and strength in pediatric patients with osteogenesis imperfecta. Bone. 2014;69:61–7.
Wronski TJ, Dann LM, Scott KS, Cintrón M. Long-term effects of ovariectomy and aging on the rat skeleton. Calcif Tissue Int. 1989;45(6):360–6.
Enderli TA, Burtch SR, Templet JN, Carriero A. Animal models of osteogenesis imperfecta: applications in clinical research. Orthop Res Rev. 2016;8:41–55.
Busse B, Bale HA, Zimmermann EA, Panganiban B, Barth HD, Carriero A, et al. Vitamin D deficiency induces early signs of aging in human bone, increasing the risk of fracture. Sci Transl Med. 2013;5(193):193ra88–8.
Zimmermann EA, Köhne T, Bale HA, Panganiban B, Gludovatz B, Zustin J, et al. Modifications to nano-and microstructural quality and the effects on mechanical integrity in Paget's disease of bone. J Bone Miner Res. 2015;30(2):264–73.
Seitz S, Priemel M, Zustin J, Beil FT, Semler J, Minne H, et al. Paget's disease of bone: histologic analysis of 754 patients. J Bone Miner Res. 2009;24(1):62–9.
Lekkala S, Taylor EA, Hunt HB, Donnelly E. Effects of diabetes on bone material properties. Curr Osteoporos Rep. 2019;17(6):455–64.
Hunt HB, Miller NA, Hemmerling KJ, Koga M, Lopez KA, Taylor EA, et al. Bone tissue composition in postmenopausal women varies with glycemic control from normal glucose tolerance to type 2 diabetes mellitus. J Bone Miner Res. 2020.
•• Daniele G, Winnier D, Mari A, Bruder J, Fourcaudot M, Pengou Z, et al. The potential role of the osteopontin–osteocalcin–osteoprotegerin triad in the pathogenesis of prediabetes in humans. Acta Diabetol. 2018;55(2):139–48 This article demonstrates that hormones playing a role in bone remodeling also affect glucose metabolism.
Creecy A, Uppuganti S, Merkel AR, O’Neal D, Makowski AJ, Granke M, et al. Changes in the fracture resistance of bone with the progression of type 2 diabetes in the ZDSD rat. Calcif Tissue Int. 2016;99(3):289–301.
Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res. 2009;24(4):597–605.
Tami AE, Nasser P, Verborgt O, Schaffler MB, Tate MLK. The role of interstitial fluid flow in the remodeling response to fatigue loading. J Bone Miner Res. 2002;17(11):2030–7.
•• McDonald MM, Khoo WH, Ng PY, Xiao Y, Zamerli J, Thatcher P, et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell. 2021;184(5):1330–1347.e13 This article demonstrates the existence of osteomorphs, new bone cells that originate trough osteoclasts fission and capable to fuse back into osteoclasts for bone repair purposes.
•• Shi C, Wu T, He Y, Zhang Y, Fu D. Recent advances in bone-targeted therapy. Pharmacol Ther. 2020;207:107473 This review presents bone-targeting moieties and strategies for the development of novel therapies applicable to bone-related pathologies.
Bessueille L, Briolay A, Como J, Mebarek S, Mansouri C, Gleizes M, et al. Tissue-nonspecific alkaline phosphatase is an anti-inflammatory nucleotidase. Bone. 2020;133:115262.
Alliston T. Biological regulation of bone quality. Curr Osteoporos Rep. 2014;12(3):366–75.
Vashishth D, Verborgt O, Divine G, Schaffler M, Fyhrie DP. Decline in osteocyte lacunar density in human cortical bone is associated with accumulation of microcracks with age. Bone. 2000;26(4):375–80.
Seref-Ferlengez Z, Kennedy OD, Schaffler MB. Bone microdamage, remodeling and bone fragility: how much damage is too much damage? BoneKEy Rep. 2015;4.
•• Alvandi LM, Chen D, Majeska RJ, Florencio-Silva R, Seref-Ferlengez Z, Schaffler MB. Mineral deposition is required to repair diffuse damage in bone in vivo. Amer Soc Bone Mineral Res; 2019. This conference proceeding abstract demonstrates new insights into the process of spontaneous repair of diffuse damage through a physicochemical remineralization process without the need of osteocytes activity.
Tsourdi E, Jähn K, Rauner M, Busse B, Bonewald LF. Physiological and pathological osteocytic osteolysis. J Musculoskelet Neuronal Interact. 2018;18(3):292.
Poulet B, Liu K, Plumb D, Vo P, Shah M, Staines K, et al. Overexpression of TIMP-3 in chondrocytes produces transient reduction in growth plate length but permanently reduces adult bone quality and quantity. PLoS One. 2016;11(12):e0167971.
Miller B, Spevak L, Lukashova L, Javaheri B, Pitsillides AA, Boskey A, et al. Altered bone mechanics, architecture and composition in the skeleton of TIMP-3-deficient mice. Calcif Tissue Int. 2017;100(6):631–40.
Kaya S, Basta-Pljakic J, Seref-Ferlengez Z, Majeska RJ, Cardoso L, Bromage TG, et al. Lactation-induced changes in the volume of osteocyte lacunar-canalicular space alter mechanical properties in cortical bone tissue. J Bone Miner Res. 2017;32(4):688–97.
Jáuregui EJ, Akil O, Acevedo C, Hall-Glenn F, Tsai BS, Bale HA, et al. Parallel mechanisms suppress cochlear bone remodeling to protect hearing. Bone. 2016;89:7–15.
• De Paolis A, Miller BJ, Doube M, Bodey AJ, Rau C, Richter C-P, et al. Increased cochlear otic capsule thickness and intracortical canal porosity in the oim mouse model of osteogenesis imperfecta. J Struct Biol. 2021:107708. This article quantifies the morphological changes of the otic capsule in mice with osteogenesis imperfecta, and how it might be involved in their hearing loss.
Brister EY, Vasi Z, Antipova O, Robinson A, Tan X, Agarwal A, et al. X-ray fluorescence microscopy: a method of measuring ion concentrations in the ear. Hear Res. 2020;391:107948.
Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016;8(5):a021873.
Zhao Y-p, Tian Q-y, Frenkel S, Liu C-j. The promotion of bone healing by progranulin, a downstream molecule of BMP-2, through interacting with TNF/TNFR signaling. Biomaterials. 2013;34(27):6412–21.
Eguchi K, Akiba Y, Akiba N, Nagasawa M, Cooper LF, Uoshima K. Insulin-like growth factor binding Protein-3 suppresses osteoblast differentiation via bone morphogenetic protein-2. Biochem Biophys Res Commun. 2018;507(1):465–70.
Turner C, Garetto L, Dunipace A, Zhang W, Wilson M, Grynpas M, et al. Fluoride treatment increased serum IGF-1, bone turnover, and bone mass, but not bone strength, in rabbits. Calcif Tissue Int. 1997;61(1):77–83.
Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, et al. Recombinant human leptin in women with hypothalamic amenorrhea. New Engl J Med. 2004;351(10):987–97.
Kawai M, Rosen CJ. The insulin-like growth factor system in bone: basic and clinical implications. Endocrinol Metab Clin N Am. 2012;41(2):323–vi.
Tang S, Alliston T. Regulation of postnatal bone homeostasis by TGFbeta. Bonekey Rep. 2013;2:255.
Balooch G, Balooch M, Nalla RK, Schilling S, Filvaroff EH, Marshall GW, et al. TGF-β regulates the mechanical properties and composition of bone matrix. ProcNat Acad Sci. 2005;102(52):18813–8.
Eckardt H, Bundgaard KG, Christensen KS, Lind M, Hansen ES, Hvid I. Effects of locally applied vascular endothelial growth factor (VEGF) and VEGF-inhibitor to the rabbit tibia during distraction osteogenesis. J Orthop Res. 2003;21(2):335–40.
Chindamo G, Sapino S, Peira E, Chirio D, Gonzalez MC, Gallarate M. Bone diseases: current approach and future perspectives in drug delivery systems for bone targeted therapeutics. Nanomaterials. 2020;10(5):875.
Paiva KBS, Granjeiro JM. Matrix Metalloproteinases in Bone Resorption, Remodeling, and Repair. Prog Mol Biol Transl Sci. 2017;148:203–303.
Shay G, Tauro M, Loiodice F, Tortorella P, Sullivan DM, Hazlehurst LA, et al. Selective inhibition of matrix metalloproteinase-2 in the multiple myeloma-bone microenvironment. Oncotarget. 2017;8(26):41827.
Nyman JS, Lynch CC, Perrien DS, Thiolloy S, O'Quinn EC, Patil CA, et al. Differential effects between the loss of MMP-2 and MMP-9 on structural and tissue-level properties of bone. J Bone Miner Res. 2011;26(6):1252–60.
Tokuhara CK, Santesso MR, Oliveira GSN, Ventura TMDS, Doyama JT, Zambuzzi WF, et al. Updating the role of matrix metalloproteinases in mineralized tissue and related diseases. J Appl Oral Sci. 2019;27:e20180596.
Paiva KB, Granjeiro JM. Bone tissue remodeling and development: focus on matrix metalloproteinase functions. Arch Biochem Biophys. 2014;561:74–87.
Tang SY, Herber RP, Ho SP, Alliston T. Matrix metalloproteinase-13 is required for osteocytic perilacunar remodeling and maintains bone fracture resistance. J Bone Miner Res. 2012;27(9):1936–50.
Lin T-H, Pajarinen J, Lu L, Nabeshima A, Cordova L, Yao Z, et al. NF-κB as a therapeutic target in inflammatory-associated bone diseases. Adv Protein Chem Struct Biol. 2017;107:117–54.
D'Oronzo S, Coleman R, Brown J, Silvestris F. Metastatic bone disease: Pathogenesis and therapeutic options: Up-date on bone metastasis management. J Bone Oncol. 2019;15:004–4.
Polyzos SA, Makras P, Tournis S, Anastasilakis AD. Off-label uses of denosumab in metabolic bone diseases. Bone. 2019;129:115048.
Jähn-Rickert K, Wölfel EM, Jobke B, Riedel C, Hellmich M, Werner M, et al. Elevated bone hardness under denosumab treatment, with persisting lower osteocyte viability during discontinuation. Front Endocrinol (Lausanne). 2020;11:250.
Plotkin LI, Bellido T. Osteocytic signalling pathways as therapeutic targets for bone fragility. Nat Rev Endocrinol. 2016;12(10):593–605.
Wang X, Yamauchi K, Mitsunaga T. A review on osteoclast diseases and osteoclastogenesis inhibitors recently developed from natural resources. Fitoterapia. 2020;142:104482.
Gourion-Arsiquaud S, Allen MR, Burr DB, Vashishth D, Tang SY, Boskey AL. Bisphosphonate treatment modifies canine bone mineral and matrix properties and their heterogeneity. Bone. 2010;46(3):666–72.
Ettinger B, Burr DB, Ritchie RO. Proposed pathogenesis for atypical femoral fractures: lessons from materials research. Bone. 2013;55(2):495–500.
Fioramonti M, Santini D, Iuliani M, Ribelli G, Manca P, Papapietro N, et al. Cabozantinib targets bone microenvironment modulating human osteoclast and osteoblast functions. Oncotarget. 2017;8(12):20113.
Nakai Y, Okamoto K, Terashima A, Ehata S, Nishida J, Imamura T, et al. Efficacy of an orally active small-molecule inhibitor of RANKL in bone metastasis. Bone Res. 2019;7:1.
Marom R, Rabenhorst BM, Morello R. Osteogenesis imperfecta: an update on clinical features and therapies. Eur J Endocrinol. 2020;183(4):R95–106.
Cosman F, Crittenden DB, Adachi JD, Binkley N, Czerwinski E, Ferrari S, et al. Romosozumab treatment in postmenopausal women with osteoporosis. New Engl J Med. 2016;375(16):1532–43.
Zhao H, Bernardo MM, Osenkowski P, Sohail A, Pei D, Nagase H, et al. Differential inhibition of membrane type 3 (MT3)-matrix metalloproteinase (MMP) and MT1-MMP by tissue inhibitor of metalloproteinase (TIMP)-2 and TIMP-3 rgulates pro-MMP-2 activation. J Biol Chem. 2004;279(10):8592–601.
Jiang C, Xia W, Wu T, Pan C, Shan H, Wang F, et al. Inhibition of microRNA-222 up-regulates TIMP3 to promotes osteogenic differentiation of MSCs from fracture rats with type 2 diabetes mellitus. J Cell Mol Med. 2020;24(1):686–94.
Sleeman A, Clements JN. Abaloparatide: A new pharmacological option for osteoporosis. Am J Health Syst Pharm. 2019;76(3):130–5.
Bernhardsson M, Aspenberg P. Abaloparatide versus teriparatide: a head to head comparison of effects on fracture healing in mouse models. Acta Orthop. 2018;89(6):674–7.
Ueland T, Stilgren L, Bollerslev J. Bone matrix levels of Dickkopf and Sclerostin are positively correlated with bone mass and strength in postmenopausal osteoporosis. Int J Mol Sci. 2019;20(12).
Ross RD, Mashiatulla M, Robling AG, Miller LM, Sumner DR. Bone matrix composition following PTH treatment is not dependent on sclerostin status. Calcif Tissue Int. 2016;98(2):149–57.
Gardinier JD, Al-Omaishi S, Rostami N, Morris MD, Kohn DH. Examining the influence of PTH(1-34) on tissue strength and composition. Bone. 2018;117:130–7.
Makowski AJ, Uppuganti S, Wadeer SA, Whitehead JM, Rowland BJ, Granke M, et al. The loss of activating transcription factor 4 (ATF4) reduces bone toughness and fracture toughness. Bone. 2014;62:1–9.
Wang X, Guo B, Li Q, Peng J, Yang Z, Wang A, et al. miR-214 targets ATF4 to inhibit bone formation. Nat Med. 2013;19(1):93–100.
Molstad DH, Mattson AM, Begun DL, Westendorf JJ, Bradley EW. Hdac3 regulates bone modeling by suppressing osteoclast responsiveness to RANKL. J Biol Chem. 2020;295(51):17713–23.
McGee-Lawrence ME, Bradley EW, Dudakovic A, Carlson SW, Ryan ZC, Kumar R, et al. Histone deacetylase 3 is required for maintenance of bone mass during aging. Bone. 2013;52(1):296–307.
Razidlo DF, Whitney TJ, Casper ME, McGee-Lawrence ME, Stensgard BA, Li X, et al. Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PLoS One. 2010;5(7):e11492.
Carpio LR, Bradley EW, McGee-Lawrence ME, Weivoda MM, Poston DD, Dudakovic A, et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci Signal. 2016;9(440):ra79–ra.
Kim J-M, Yang Y-S, Park KH, Ge X, Xu R, Li N, et al. A RUNX2 stabilization pathway mediates physiologic and pathologic bone formation. Nat Commun 2020;11(1):1–17.
Valenti MT, Mottes M, Cheri S, Deiana M, Micheletti V, Cosaro E, et al. Runx2 overexpression compromises bone quality in acromegalic patients. Endocr Relat Cancer. 2018;25(3):269–77.
Kim W-J, Shin H-L, Kim B-S, Kim H-J, Ryoo H-M. RUNX2-modifying enzymes: therapeutic targets for bone diseases. Exp Mol Med. 2020;52(8):1178–84.
Schroeder TM, Westendorf JJ. Histone deacetylase inhibitors promote osteoblast maturation. J Bone Miner Res. 2005;20(12):2254–63.
Ouyang N, Li H, Wang M, Shen H, Si J, Shen G. The transcription factor Foxc1 promotes osteogenesis by directly regulating Runx2 in response of intermittent parathyroid hormone (1–34) treatment. Front Pharmacol. 2020;11:592.
Besio R, Garibaldi N, Leoni L, Cipolla L, Sabbioneda S, Biggiogera M, et al. Cellular stress due to impairment of collagen prolyl hydroxylation complex is rescued by the chaperone 4-phenylbutyrate. Dis Model Mech. 2019;12(6).
Besio R, Iula G, Garibaldi N, Cipolla L, Sabbioneda S, Biggiogera M, et al. 4-PBA ameliorates cellular homeostasis in fibroblasts from osteogenesis imperfecta patients by enhancing autophagy and stimulating protein secretion. Biochimica et Biophysica Acta (BBA)-Molecul Basis Dis. 2018;1864(5):1642–52.
Ambrosi TH, Scialdone A, Graja A, Gohlke S, Jank A-M, Bocian C, et al. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell. 2017;20(6):771–84 e6.
Yang Y, Zhao C, Liang J, Yu M, Qu X. Effect of dipeptidyl peptidase-4 inhibitors on bone metabolism and the possible underlying mechanisms. Front Pharmacol. 2017;8:487.
Yoshida T, Wang J, Stern PH. Gonadal hormones and bone. Handb Exp Pharmacol. 2020;262:65–91.
Powell KM, Skaggs C, Pulliam A, Berman A, Allen MR, Wallace JM. Zoledronate and Raloxifene combination therapy enhances material and mechanical properties of diseased mouse bone. Bone. 2019;127:199–206.
Allen MR, Hogan HA, Hobbs WA, Koivuniemi AS, Koivuniemi MC, Burr DB. Raloxifene enhances material-level mechanical properties of femoral cortical and trabecular bone. Endocrinology. 2007;148(8):3908–13.
Yoon SH, Grynpas M, Mitchell J. Intermittent PTH treatment improves bone and muscle in glucocorticoid treated Mdx mice: a model of duchenne muscular dystrophy. Bone. 2019;121:232–42.
Chiang CY, Zebaze RMD, Ghasem-Zadeh A, Iuliano-Burns S, Hardidge A, Seeman E. Teriparatide improves bone quality and healing of atypical femoral fractures associated with bisphosphonate therapy. Bone. 2013;52(1):360–5.
Varela A, Chouinard L, Lesage E, Guldberg R, Smith SY, Kostenuik PJ, et al. One year of abaloparatide, a selective peptide activator of the PTH1 receptor, increased bone mass and strength in ovariectomized rats. Bone. 2017;95:143–50.
Javaheri B, Carriero A, Staines KA, Chang Y-M, Houston D, Oldknow KJ, et al. Phospho1 deficiency transiently modifies bone architecture yet produces consistent modification in osteocyte differentiation and vascular porosity with ageing. Bone. 2015;81:277–91.
Carriero A, Bruse JL, Oldknow KJ, Millán JL, Farquharson C, Shefelbine SJ. Reference point indentation is not indicative of whole mouse bone measures of stress intensity fracture toughness. Bone. 2014;69:174–9.
Kiffer-Moreira T, Yadav MC, Zhu D, Narisawa S, Sheen C, Stec B, et al. Pharmacological inhibition of PHOSPHO1 suppresses vascular smooth muscle cell calcification. J Bone Miner Res. 2013;28(1):81–91.
Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol-Cell Physiol. 2001;281(1):C1–C11.
Rodriguez-Florez N, Carriero A, Shefelbine SJ. The use of XFEM to assess the influence of intra-cortical porosity on crack propagation. Comput Meth Biomechan Biomed Eng. 2017;20(4):385–92.
Cipriani C, Colangelo L, Santori R, Renella M, Mastrantonio M, Minisola S, et al. The interplay between bone and glucose metabolism. Front Endocrinol. 2020;11:122.
Mu C-F, Shen J, Liang J, Zheng H-S, Xiong Y, Wei Y-H, et al. Targeted drug delivery for tumor therapy inside the bone marrow. Biomaterials. 2018;155:191–202.
Skjødt MK, Frost M, Abrahamsen B. Side effects of drugs for osteoporosis and metastatic bone disease. Br J Clin Pharmacol. 2019;85(6):1063–71.
Feng Q, Xu J, Zhang K, Yao H, Zheng N, Zheng L, et al. Dynamic and cell-infiltratable hydrogels as injectable carrier of therapeutic cells and drugs for treating challenging bone defects. ACS Cent Sci. 2019;5(3):440–50.
Cai M, Yang L, Zhang S, Liu J, Sun Y, Wang X. A bone-resorption surface-targeting nanoparticle to deliver anti-miR214 for osteoporosis therapy. Int J Nanomedicine. 2017;12:7469.
Lavrador P, Gaspar VM, Mano JF. Stimuli-responsive nanocarriers for delivery of bone therapeutics–Barriers and progresses. J Control Release. 2018;273:51–67.
Acknowledgements
This work was supported by the National Science Foundation (CBET-1829310).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors state that they have no conflicts of interest.
Human and animal rights
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Biomechanics
Rights and permissions
About this article

Cite this article
Muñoz, A., Docaj, A., Ugarteburu, M. et al. Poor bone matrix quality: What can be done about it?. Curr Osteoporos Rep 19, 510–531 (2021). https://doi.org/10.1007/s11914-021-00696-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-021-00696-6