
Abstract
The importance of the insulin-like growth factor (IGF)-I axis in the regulation of bone size and bone mineral density, two important determinants of bone strength, has been well established from clinical studies involving patients with growth hormone deficiency and IGF-I gene disruption. Data from transgenic animal studies involving disruption and overexpression of components of the IGF-I axis also provide support for a key role for IGF-I in bone metabolism. IGF-I actions in bone are subject to regulation by systemic hormones, local growth factors, as well as mechanical stress. In this review we describe findings from various genetic mouse models that pertain to the role of endocrine and local sources of IGF-I in the regulation of skeletal growth.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the key functions of bone is to provide mechanical support and protection for the organism, and, therefore, bones must continue to grow both in length and size until adulthood. The changes in bone length and width are driven by cells of chondrocytic lineage in the growth plate and osteoblastic lineage cells in the periosteum, respectively [1–3]. Longitudinal bone growth occurs at the growth plate whereby an anlage of cartilage is gradually replaced by mineralized tissue by a process called endochondral ossification [1–3]. Bone growth in width occurs by periosteal apposition owing to the action of periosteal osteoblasts. The changes in both length and size contribute to the large increase in bone mass that occurs during childhood and puberty [4–6]. Bone mass continues to increase after puberty, albeit gradually, until peak bone mass is reached around 20–30 years of age, and then gradually declines in aging men and women [4–6]. The achievement of an optimal peak bone mass is recognized as a primary health goal during early growth periods since it is widely understood that the risk of fragility fractures in old age has its origin during growth [7, 8]. It is known that the marked changes in bone mass during childhood and puberty are governed by a complex interplay of hormones, local growth factors, nutritional factors, and mechanical forces [1, 3, 9–11]. Of the various regulatory molecules that govern skeletal growth, it is now widely accepted that insulin-like growth factor (IGF)-I is a critically important factor in regulating bone growth longitudinally as well as in width in both humans and in experimental animals [11–15]. In this chapter, we discuss regulation and mechanisms of IGF-I action during growth with particular emphasis on areas in which recent advances have been made.
Regulators of IGF-I Expression
Since the discovery that sulfation factor/somatomedin-C/IGF-I is the major mediator of growth hormone (GH) action, it has become abundantly clear that IGF-I plays a central role in growth, development, and metabolism of skeletal tissues [15, 16]. According to the original somatomedin hypothesis, GH has been proposed to stimulate skeletal growth indirectly by stimulating liver production of IGF-I to act in an endocrine manner to stimulate bone growth. However, subsequent studies have shown that GH also has direct effects on bone and that these effects are largely mediated via GH regulation of local IGF-I expression and its action in bone [12, 17, 18]. Accordingly, both chondrocytes and osteoblasts contain GH receptors and GH treatment increases the production of IGF-I in both of these cell types [12, 17, 18]. Genetic mouse models have been used to determine the relative contribution of liver-derived versus locally produced IGF-I in mediating GH effects on bone [12, 15]. Fan et al. [19] have recently shown that deletion of GH receptor in liver had no effect on total body or bone linear growth despite a greater than 90 % suppression of circulating IGF-I, although trabecular bone volume was significantly decreased in mice lacking the GH receptor in the liver. In another study, it was found that the GH effect on osteoblast number both in vitro and in vivo was dependent on local production of IGF-I [20]. These data suggest that IGF-I is indeed a major regulator of IGF-I action and that GH effects on bone are mediated via GH-induced IGF-I production in both liver and bone.
It is now known that IGF-I expression in bone is regulated by GH as well as many systemic and local regulators of bone growth. In this regard, our recent studies have shown that thyroid hormone (TH) is a key regulator of IGF-I expression, particularly during the prepubertal growth period [21••]. Studies using genetic mouse models deficient in TH revealed that serum levels of IGF-I were reduced by more than 50 % at day 21 compared to wild-type mice as a consequence of a decrease in IGF-I expression in liver and bone. TH treatment for 10 days during the prepubertal growth period increased IGF-I expression in both liver and bone and normalized the serum IGF-I levels [21••]. Consistent with the mouse data, clinical studies in humans have shown that the elevated levels of serum IGF-I in hyperthyroid patients were brought back to normal levels by treatment with methimazole to correct the TH levels [22]. In terms of target cell types for TH effects on IGF-I expression, we and others have found that TH treatment increases IGF-I expression in bone cells and chondrocytes [21••, 23, 24]. Furthermore, in vitro studies revealed that TH, in the presence of TH receptor-α, bound to the TH response element in intron 1 of the IGF-I gene to stimulate transcription [21••]. Thus, TH is a key regulator of both local and endocrine IGF-I action during the prepubertal growth period.
The increase in IGF-I expression during the pubertal growth period when substantial periosteal expansion and bone mass accrual are attained is generally assumed to be mediated via sex hormones. Both androgens and estrogens can stimulate IGF-I secretion [25] and increase pulsatility of GH secretion, characteristic of puberty [26], with testosterone’s effect being dependent on aromatization of estrogen. In males, the androgen-induced increase in GH correlated with an increase in IGF-I. In females, an estrogen-induced increase in GH is sometimes accompanied by a decrease in IGF-I. This phenomenon appears to be dose-dependent, with higher doses being inhibitory of IGF-I release, and can be dependent on route of administration [27, 28]. The mechanisms by which androgens and estrogens interact with GH to modulate transcriptional regulation of the GH response genes remain to be established.
Studies on potential mediators of parathyroid hormone (PTH) anabolic action have revealed that IGF-I is required for the bone-forming effects of PTH. In this regard, earlier studies revealed that PTH stimulates IGF-I production in osteoblasts and that IGF-I can reproduce the effects of PTH on osteoblast proliferation, differentiation, and survival [29–31]. Furthermore, PTH effects on collagen synthesis, alkaline phosphatase activity, as well as the expression of osteocalcin were blocked by the addition of IGF-I–neutralizing antibodies, thus suggesting that the PTH effect on osteoblasts is dependent on local production of IGF-I [31, 32]. Consistent with these in vitro data, studies using genetic mouse models with disruption of IGF-I [33, 34] or insulin receptor substrate-1 [35] have provided irrevocable direct evidence that PTH effects on bone formation in vivo are also dependent on osteoblast production of IGF-I. Glucocorticoid represents another example of IGF-I regulation in bone by a systemic regulator. Glucocorticoid treatment has been shown to reduce mean wall thickness of trabecular bone, which reflects the total amount of bone formed during a remodeling cycle [3, 29, 36]. In vitro evidence shows that glucocorticoid decreases both the proliferative and differentiated functions of osteoblasts and is associated with a decrease in production of IGF-I and its stimulatory binding protein [36–38]. In addition, Jux et al. [39] have shown that glucocorticoid decreases both basal and IGF-induced chondrocyte replication and that these effects are prevented by the addition of IGF-I–neutralizing antibodies.
In addition to systemic regulators, IGF-I expression in osteoblasts has also been shown to be regulated by locally produced growth factors including fibroblast growth factor 2, transforming growth factor-β1, bone morphogenetic protein 7, and interleukin-1 [40–44]. In addition to systemic and local regulators, another key regulator of IGF-I expression in bone is mechanical strain (see below). Thus, the finding that a number of bone formation factors regulate IGF-I expression is consistent with the idea that IGFs play a central role in the regulation of bone formation and that a number of systemic and local regulators mediate their bone-forming effects in part via modulating local production of IGF-I.
Effects of IGF-I on the Skeleton
Studies by Baker et al. [45] and Liu et al. [46] have shown that while IGF-II is essential for normal embryonic growth, IGF-I has a continuous growth-promoting function to regulate growth of various tissues throughout development. Accordingly, our studies have shown that mice with targeted disruption of IGF-I exhibit 40 % reduced femoral length, 38 % reduced bone size, and 87 % reduced bone mineral content at the femur (Table 1) [47]. Femoral areal bone mineral density (BMD) and volumetric BMD were reduced by 56 % and 32 %, respectively, in the IGF-I knockout mice compared to corresponding control littermate mice at 8 weeks of age. Consistent with the notion that GH is a major regulator of IGF-I action, it has been shown that mice with disruption of GH-releasing hormone receptor or GH receptor functions exhibit reduced bone length and bone size [48]. Comparison of skeletal deficits in IGF-I knockout and GH-deficient mice reveal that both GH-dependent and GH-independent mechanisms contribute to peak bone mass and that much of the GH-independent effect on the skeleton is mediated during the prepubertal growth period [21••, 47, 49].
Histomorphometric studies revealed that IGF-I knockout mice had bone formation [50] and mineral apposition rates that were approximately 25 % and 50 % of the wild-type mice, respectively, at the periosteum of the tibiofibular junction. In contrast to the cortical bone, trabecular bone volume of the proximal tibia but not lumbar vertebra was increased in the IGF-I knockout mice compared to wild-type mice due to increased trabecular connectivity, number, and decreased spacing [50]. These data suggest that the effect of IGF-I on bone structure may be complex depending on the compartment (cortical vs trabecular) as well as the region (appendicular vs axial).
Consistent with the mouse data, patients with mutations in the IGF-I receptor have been shown to exhibit varying degrees of intrauterine and postnatal growth retardation [14, 51, 52]. Walenkamp et al. [52] have reported that a 55-year-old patient with a missense mutation in the IGF-I gene that results in the substitution of valine at position 44 with methionine has 4–5 standard deviation reduced BMD at both femoral neck and lumbar spine compared to age-matched normal subjects. The IGF-I mutant protein produced in this patient has a 100-fold reduced affinity for IGF-I receptor compared to native IGF-I. Thus, there is convincing evidence that IGF-I plays an important role in the regulation of peak bone mass in both humans and experimental animals.
Endocrine IGF-I Action
IGF-I circulates in high abundance in the blood, which raises an obvious question on the need for high concentrations of IGFs in the blood. About 75 % of the IGF-I is complexed with insulin-like growth factor-binding protein (IGFBP)-3 and an acid-labile subunit (ALS) in a 150- to 200-kDa ternary complex, whereas the remainder of the circulating IGF-I is bound to lower molecular mass IGFBPs [53]. Only a small fraction (<1 %) of IGFs exists in free form in the blood. Since circulating levels of IGF-I are largely determined by GH and nutrition, this suggests that endocrine IGF-I may function to promote general growth. To determine the role of endocrine IGF-I to bone growth, several elegant mouse models with liver-specific manipulation of IGF-I and/or other IGF system components have been developed during the past 12 years. In 1999, two mouse models with liver-specific IGF-I inactivation were developed [54, 55]. Both models had exon 4 of the IGF-I gene inactivated in hepatocytes using albumin-Cre–mediated or an inducible Mx1-Cre–mediated DNA excision models. These studies revealed that the skeletal deficit caused by disruption of liver-produced IGF-I is rather small despite a greater than 75 % reduction in circulating levels of IGF-I.
One potential explanation for the lack of substantial skeletal phenotype in mice with disruption of liver IGF-I is that the remaining 25 % of circulating levels of IGF-I is more bioavailable and sufficient to maintain skeletal growth. An alternate possibility is that disruption of the IGF-I gene in the liver became effective only after a critical postweaning period of growth spurt in these genetic mouse models. To rule out these possibilities, Yakar et al. [56••, 57] generated double knockout mice with disruption of liver-derived IGF-I and total ALS and triple knockout mice with disruption of liver-derived IGF-I, total ALS, and total IGFBP-3. The double and triple knockout mouse models exhibited a 90 % and 97.5 % reduction in serum IGF-I levels, respectively, compared to corresponding wild-type mice. The triple knockout mice exhibited a modest reduction in total body length compared to total IGF-I knockout mice, thus suggesting a relatively minor contribution of circulating IGF-I in regulating longitudinal bone growth. On the other hand, endocrine IGF-I appears to contribute considerably to the periosteal expansion, as the reduction in cortical bone width in the triple knockout mice was nearly 50 % of that seen in the total IGF-I knockout mice [47]. The findings that the substantial proportion of changes in bone size but not length during embryonic and postnatal growth is dependent on endocrine IGF-I action suggest that the mechanisms by which IGF-I regulates growth in length versus width may be different.
In another study, Stratikopoulos et al. [58] generated bitransgenic mice by crossing IGF-I floxed mice in which a dormant IGF-I cDNA was placed downstream of a transcriptional “stop” DNA sequence flanked by loxP sites in an IGF-I null background with cre transgenic mice in which cre expression was under the control of a liver-specific human α1-antitrypsin gene promoter. In this model, the IGF-I cDNA was inserted by knock-in into the mutated and inactive IGF-I locus itself to ensure proper transcriptional regulation. This study also demonstrated that approximately 30 % of the body growth could be achieved by liver-specific IGF-I re-expression. Based on these data, it can be concluded that while liver-derived IGF-I can function as a bona fide hormone to enhance bone growth in the absence of local IGF-I, both endocrine and local IGF-I action are needed for normal bone growth.
One major confounder that is common to all genetic mouse models of deficient endocrine IGF-I action is that the reduction in circulating IGF-I in these models disrupts a negative feedback pathway such that GH secretion from the anterior pituitary is unregulated. The interpretation of data from these models became complicated since the elevated GH levels could increase local IGF-I expression as well as exert IGF-I–independent effects on target tissues such as bone. To overcome this drawback, Nordstrom et al. [59•] generated compound mutant (Lit-JAK2L) mice by crossing GH-deficient GH-releasing hormone receptor mutant (Lit) mice with mice with hepatocyte-specific disruption of Janus kinase 2 (JAK2), a protein that is essential for GH-stimulated IGF-I production. Upon treatment of compound mutant mice (Lit-JAK2L) and corresponding control mice (Lit-Con) with equal amounts of GH such that the only difference between the two groups was hepatic GH signaling, the authors found that the GH-mediated acquisition of BMD was reduced in Lit-JAK2L mice compared to Lit-Con mice. While these data are consistent with a role for hepatic IGF-I in regulating skeletal growth, one disadvantage with this model is the recognition that JAK2 is involved in mediating the effects of other cytokines besides GH.
Local IGF-I Action
In addition to IGF-I in the circulation, IGF-I is also available to skeletal tissues through de novo synthesis by various cell types present in bone and also by release of IGF-I from bone matrix during osteoclastic bone resorption [42]. It has been shown previously that IGFs are the most abundant growth factors produced by bone cells in vitro and stored in bone matrix. Neutralization of locally produced IGFs in serum-free cultures resulted in nearly 50 % inhibition of basal osteoblast proliferation, thus suggesting that locally produced IGFs contribute substantially to basal bone cell proliferation [60]. Furthermore, as stated earlier, osteoblast cell production of IGF-I is known to be regulated both by systemic and local regulators of bone formation, suggesting potential involvement of IGF-I in mediating the effects of systemic hormones and local growth factors.
To evaluate the role of locally produced IGF-I in the regulation of bone formation in vivo, both transgenic overexpression and conditional knockout mouse models have been used. Zhao et al. [61] generated transgenic mice that overexpressed IGF-I specifically in mature osteoblasts by driving transgene expression using an osteocalcin-specific promoter. Targeted overexpression of IGF-I to osteoblasts of transgenic mice increased the cancellous bone formation rate and volume without any change in osteoblast number, suggesting that locally delivered IGF-I exerts anabolic effects primarily via increasing the activity of resident osteoblasts. Consistent with these data, Jiang et al. [62] demonstrated that transgenic overexpression of IGF-I in cells of osteoblast lineage using 3.6 kb of 5′ upstream regulatory sequence, and most of the first intron of the rat type I collagen α1 gene, resulted in increased indices of both bone formation and resorption. Furthermore, transgenic mouse models involving overexpression of components of the IGF system (eg, IGFBPs −4 and −5, and IGFBP proteases) that modulate IGF bioavailability also provide evidence that locally produced IGF-I participates in bone formation [61, 63–67].
Although the transgenic studies provide evidence for the involvement of locally produced IGF-I in regulating osteoblast functions, one limitation with transgenic approaches is that the levels of transgene expressed are often too high to mimic physiological conditions. To overcome this potential drawback, we and others have used the Cre/loxP approach to specifically disrupt the IGF-I or IGF-I receptor in various bone cell types. Zhang et al. [68] generated mice with conditional disruption of IGF-I receptor in mature osteoblasts by crossing IGF-I receptor loxP mice with Cre mice in which Cre expression was driven by the human osteocalcin promoter. Mice with conditional deletion of the IGF-I receptor in mature osteoblasts showed significant reduction in distal metaphyseal trabecular bone volume, trabecular thickness, trabecular number and mineral apposition rate, as well as increased osteoid volume, implying a key role for IGF-I in mineralization. Because disruption of IGF-I receptor in osteoblasts would impair not only the autocrine/paracrine IGF-I actions but also endocrine IGF-I effects, this model did not distinguish the role of local versus endocrine IGF-I in regulating bone formation in vivo.
To study the relative contribution of IGF-I produced by cells of the osteoblast lineage in regulating skeletal growth, we disrupted IGF-I expression in type I collagen-producing osteoblast cells by crossing IGF-I floxed Cre mice in which Cre expression was driven by the entire collagen type 1 α2 promoter. Expression of IGF-I was decreased in the bones but not in the livers of conditional knockout mice [69]. Accordingly, circulating serum levels of IGF-I were unaffected by conditional disruption of IGF-I in type I collagen-producing cells. Disruption of local IGF-I caused dramatic reductions in BMD and bone size. Histomorphometric studies revealed significant decreases in the bone formation rate and mineral apposition rate in the conditional mutants, thus suggesting that local IGF-I deficiency resulted in impaired differentiation and/or function of osteoblasts [69]. Thus, studies using mice with conditional knockout of IGF-I or the IGF-I receptor in osteoblasts support the notion that locally produced IGF-I is critical for optimal skeletal development and subsequent mineralization.
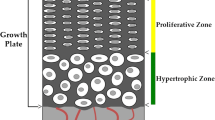
In addition to cells of the osteoblast lineage, chondrocytes represent another major cell type in bone that expresses IGF-I. We have reported that mice with conditional disruption of IGF-I in chondrocytes exhibited reduced bone length, total body areal BMD, and bone width [70]. The reduced expression levels of parathyroid hormone–related protein (PTHrP), Dlx-5, and Sox-9 in the bones of conditional mutants suggest that IGF-I produced by chondrocytes may regulate longitudinal growth and bone width, in part via regulating expression of one or more growth factors involved in chondrocyte proliferation/differentiation. Since circulating IGF-I levels were not altered in mice with conditional disruption of the IGF-1 gene in chondrocytes, it implies a local role for chondrocyte-produced IGF-I in regulating longitudinal bone growth but also bone width and bone mass accrual. Consistent with these data, Wang et al. [71] have recently reported that tamoxifen-inducible cartilage-specific IGF-I receptor knockout mice exhibited growth retardation with a disorganized growth plate and reduced chondrocyte proliferation. Surprisingly, PTHrP expression was increased in the growth plates of cartilage-specific IGF-I receptor knockout mice. In any case, studies on mouse models with disruption of cartilage-specific IGF-I or IGF-I receptor are consistent with a role for chondrocyte-produced IGF-I in regulating proliferation, survival, and differentiation of chondrocytes in the growth plate.
Mechanical Loading and IGF-I
Mechanical loading causes a rapid induction of IGF-I mRNA levels within 4 h after loading in the loaded tibia compared to the contralateral unloaded bone [72–75]. In a second study, a single 10-minute episode of mechanical stimulation increased IGF-I expression in osteocytes of loaded bone within 6 h after loading [73]. In vitro studies using monolayer cultures of TE85 cells and human primary osteoblasts have shown that the osteoblast proliferation responses to mechanical strain and estrogen are mediated via the estrogen receptor-α and the IGF-I receptor [76–78]. Consistent with a role for IGF-I in mediating a mechanical strain response in bone are the in vivo findings that transgenic mice selectively overexpressing IGF-I in osteoblasts exhibit a fivefold increase in the periosteal bone formation response to a low-magnitude loading regimen [79]. In contrast, there was no significant increase in periosteal bone formation response to mechanical loading in wild-type mice, thus suggesting that IGF-I and mechanical strain produce a synergistic response to stimulating periosteal bone formation. In another study, it was reported that IGF-I treatment induced bone formation in GH-deficient normally loaded rats but not in unloaded rats, thus suggesting skeletal unloading induces resistance to IGF-I [80, 81]. Studies on the mechanism by which mechanical strain and IGF-I interact to regulate the number and activity of osteoblast lineage cells revealed evidence for an integrin-dependent upregulation of IGF-I receptor phosphorylation and recruitment of SHP1 and/or SHP2 to IGF-I receptor and inhibition of SHP-mediated IGF-I receptor phosphorylation [82, 83]. More recently, Long et al. [84••] have shown that skeletal unloading-induced resistance to IGF-I involving integrins is selective to IGF-I and not shared by platelet-derived growth factor (PDGF).
Although there was a considerable body of data in the literature to implicate a role for IGF-I in mediating the mechanical strain response in bone, direct evidence to demonstrate a cause and effect relationship between an increase in IGF-I expression and skeletal changes was lacking. To test a causal role for IGF-I in the bone anabolic response to mechanical loading, we generated conditional IGF-I knockout mice using a Cre-loxP approach in which IGF-I expression was disrupted in type I collagen-producing osteoblasts and subjected these mice to mechanical loading. We found that 2 weeks of 4-point bending caused a significant increase in the periosteal bone formation response in wild-type mice and not in IGF-I conditional knockout mice, thus suggesting that mechanical loading-induced periosteal bone expansion is dependent on local IGF-I production in bone [85••]. Furthermore, axial loading produced an 8 % to 25 % increase in trabecular parameters (eg, bone volume, thickness, bone density) at the secondary spongiosa of wild-type but not IGF-I knockout mice. Consistent with this in vivo data, blockade of IGF-I action with inhibitory IGFBP-4 in vitro completely abolished fluid-flow stress-induced osteoblast cell proliferation. Studies on expression levels of genes that are influenced by the mechanical strain response revealed that several genes in the ephrin signaling pathway were found to be upregulated by mechanical loading in the wild-type mice but not in the IGF-I conditional knockout mice [85••], thus suggesting that IGF-I is upstream of ephrin signaling in the mechanical loading signaling pathway. These studies show that IGF-I expressed in type 1 collagen-producing bone cells is critical for converting the mechanical signal into an anabolic signal in bone, and other growth factors cannot compensate for the loss of local IGF-I.
Conclusions
Studies using various genetic mouse models as well as human clinical studies have provided irrevocable evidence for a key role for IGF-I in the regulation of skeletal growth throughout the entire lifespan. While studies using a Cre/loxP approach to disrupt IGF-I specifically in various cell types have shown that both endocrine and local IGF-I actions are necessary for optimal skeletal growth (Fig. 1), the exact contribution of the endocrine and local sources of IGF-I have not been teased out because of lack of specificity of the Cre models in specifically disrupting IGF-I gene expression in a cell type of interest. Studies also have demonstrated that IGF-I plays an essential role in mediating not only the effects of GH but also other key systemic and local regulators of bone formation. The availability of the various genetic mouse models will provide an opportunity to address a number of unanswered questions that relate to the mechanism and role of IGF-I: 1) Do endocrine and local IGF-I regulate skeletal growth via similar or distinct mechanisms? 2) Is endocrine IGF-I action more important than local IGF-I action or vice versa during specific growth periods (embryonic, prepubertal, pubertal, postpubertal)? 3) Is loss of bone maintenance with age predominantly due to deficiency in local and/or endocrine IGF-I action? 4) What is the role of IGF-I produced by osteoclasts and osteocytes? 5) Are IGF-I biological effects in various bone cell types mediated via similar or distinct signaling pathways? 6) Is manipulation of local or endocrine IGF-I action more effective in promoting bone anabolism?

Model of growth hormone regulation of skeletal growth involving modulation of endocrine and local insulin-like growth factor-I (IGF-I) actions. Growth hormone increases liver production of IGF-I, which is transported to bone via blood to act as an endocrine hormone. In the blood, the majority of IGF-I exists as a 150-kDa complex consisting of acid-labile subunit (ALS)+IGF-binding protein (IGFBP)3+IGF-I. Approximately 25 % of IGF-I exists as a 40- to 50-kDa IGFBP+IGF-I complex. Only a minor fraction of IGF-I (<1 %) exists in free form. In addition, growth hormone is also known to increase production of IGF-I in bone cells whereby it acts as a local growth factor in an autocrine/paracrine manner to stimulate growth
References
Papers of particular interest, published recently, have been highlighted as:• Of importance •• Of major importance
Lefebvre V, Bhattaram P. Vertebrate skeletogenesis. Curr Top Dev Biol. 2010;90:291–317.
Olsen BR, Reginato AM, Wang W. Bone development. Annu Rev Cell Dev Biol. 2000;16:191–220.
van der Eerden BC, Karperien M, Wit JM. Systemic and local regulation of the growth plate. Endocr Rev. 2003;24:782–801.
Osteoporosis prevention, diagnosis, and therapy. NIH Consens Statement 2000; 17:1–45.
Heaney RP, Abrams S, Dawson-Hughes B, Looker A, Marcus R, Matkovic V, Weaver C. Peak bone mass. Osteoporos Int. 2000;11:985–1009.
Libanati C, Baylink DJ, Lois-Wenzel E, Srinvasan N, Mohan S. Studies on the potential mediators of skeletal changes occurring during puberty in girls. J Clin Endocrinol Metab. 1999;84:2807–14.
Gafni RI, Baron J. Childhood bone mass acquisition and peak bone mass may not be important determinants of bone mass in late adulthood. Pediatrics. 2007;119 Suppl 2:S131–6.
Hansen MA, Overgaard K, Riis BJ, Christiansen C. Role of peak bone mass and bone loss in postmenopausal osteoporosis: 12 year study. BMJ. 1991;303:961–4.
de Crombrugghe B, Lefebvre V, Behringer RR, Bi W, Murakami S, Huang W. Transcriptional mechanisms of chondrocyte differentiation. Matrix Biol. 2000;19:389–94.
Lefebvre V, Smits P. Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today. 2005;75:200–12.
Mackie EJ, Tatarczuch L, Mirams M. The growth plate chondrocyte and endochondral ossification. J. Endocrinol. In Press; 2011.
Ohlsson C, Mohan S, Sjogren K, Tivesten A, Isgaard J, Isaksson O, Jansson JO, Svensson J. The role of liver-derived insulin-like growth factor-I. Endocr Rev. 2009;30:494–535.
Olson LE, Ohlsson C, Mohan S. The role of GH/IGF-I-mediated mechanisms in sex differences in cortical bone size in mice. Calcif Tissue Int. 2011;88:1–8.
Walenkamp MJ, Wit JM. Genetic disorders in the GH IGF-I axis in mouse and man. Eur J Endocrinol. 2007;157 Suppl 1:S15–26.
Yakar S, Courtland HW, Clemmons D. IGF-1 and bone: New discoveries from mouse models. J Bone Miner Res. 2010;25:2543–52.
LeRoith D. Clinical relevance of systemic and local IGF-I: lessons from animal models. Pediatr Endocrinol Rev. 2008;5 Suppl 2:739–43.
Mohan S, Baylink DJ. Role of growth hormone/insulin-like growth factor axis. In: Glowacki J, Rosen CJ, Bilezikian JP, editors. The aging skeleton. San Diego: Academic; 1999. p. 209–19.
Ohlsson C, Bengtsson BA, Isaksson OG, Andreassen TT, Slootweg MC. Growth hormone and bone. Endocr Rev. 1998;19:55–79.
Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ, Kopchick JJ, Le Roith D, Trucco M, Sperling MA. Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J Biol Chem. 2009;284:19937–44.
DiGirolamo DJ, Mukherjee A, Fulzele K, Gan Y, Cao X, Frank SJ, Clemens TL. Mode of growth hormone action in osteoblasts. J Biol Chem. 2007;282:31666–74.
•• Xing W, Govoni K, Donahue LR, Kesavan C, Wergedal J, Long C, Bassett JH, Gogakos A, Wojcicka A, Williams GR, et al. Genetic evidence that thyroid hormone is indispensable for prepubertal IGF-I expression and bone acquisition in mice. J. Bone Miner Res. 2012; 27:1067–79. Reports that TH acting via IGF-dependent and -independent mechanisms plays a more critical role than GH in the regulation of skeletal growth during the prepubertal growth period.
Lakatos P, Foldes J, Nagy Z, Takacs I, Speer G, Horvath C, Mohan S, Baylink DJ, Stern PH. Serum insulin-like growth factor-I, insulin-like growth factor binding proteins, and bone mineral content in hyperthyroidism. Thyroid. 2000;10:417–23.
Lakatos P, Caplice MD, Khanna V, Stern PH. Thyroid hormones increase insulin-like growth factor I content in the medium of rat bone tissue. J Bone Miner Res. 1993;8:1475–81.
O’Shea PJ, Bassett JH, Sriskantharajah S, Ying H, Cheng SY, Williams GR. Contrasting skeletal phenotypes in mice with an identical mutation targeted to thyroid hormone receptor alpha1 or beta. Mol Endocrinol. 2005;19:3045–59.
Styne DM. The regulation of pubertal growth. Horm Res. 2003;60:22–6.
Veldhuis JD, Metzger DL, Martha Jr PM, Mauras N, Kerrigan JR, Keenan B, Rogol AD, Pincus SM. Estrogen and testosterone, but not a nonaromatizable androgen, direct network integration of the hypothalamo-somatotrope (growth hormone)-insulin-like growth factor I axis in the human: evidence from pubertal pathophysiology and sex-steroid hormone replacement. J Clin Endocrinol Metab. 1997;82:3414–20.
Leung KC, Johannsson G, Leong GM, Ho KK. Estrogen regulation of growth hormone action. Endocr Rev. 2004;25:693–721.
Meinhardt UJ, Ho KK. Modulation of growth hormone action by sex steroids. Clin Endocrinol (Oxf). 2006;65:413–22.
Canalis E. Insulin like growth factors and the local regulation of bone formation. Bone. 1993;14:273–6.
Linkhart TA, Mohan S. Parathyroid hormone stimulates release of insulin-like growth factor-I (IGF-I) and IGF-II from neonatal mouse calvaria in organ culture. Endocrinology. 1989;125:1484–91.
McCarthy TL, Centrella M, Canalis E. Parathyroid hormone enhances the transcript and polypeptide levels of insulin-like growth factor I in osteoblast-enriched cultures from fetal rat bone. Endocrinology. 1989;124:1247–53.
Ishizuya T, Yokose S, Hori M, Noda T, Suda T, Yoshiki S, Yamaguchi A. Parathyroid hormone exerts disparate effects on osteoblast differentiation depending on exposure time in rat osteoblastic cells. J Clin Invest. 1997;99:2961–70.
Bikle DD, Sakata T, Leary C, Elalieh H, Ginzinger D, Rosen CJ, Beamer W, Majumdar S, Halloran BP. Insulin-like growth factor I is required for the anabolic actions of parathyroid hormone on mouse bone. J Bone Miner Res. 2002;17:1570–8.
Miyakoshi N, Kasukawa Y, Linkhart TA, Baylink DJ, Mohan S. Evidence that anabolic effects of PTH on bone require IGF-I in growing mice. Endocrinology. 2001;142:4349–56.
Yamaguchi M, Ogata N, Shinoda Y, Akune T, Kamekura S, Terauchi Y, Kadowaki T, Hoshi K, Chung UI, Nakamura K, et al. Insulin receptor substrate-1 is required for bone anabolic function of parathyroid hormone in mice. Endocrinology. 2005;146:2620–8.
Canalis E. Mechanisms of glucocorticoid action in bone. Curr Osteoporos Rep. 2005;3:98–102.
Cheng SL, Zhang SF, Mohan S, Lecanda F, Fausto A, Hunt AH, Canalis E, Avioli LV. Regulation of insulin-like growth factors I and II and their binding proteins in human bone marrow stromal cells by dexamethasone. J Cell Biochem. 1998;71:449–58.
Chevalley T, Strong DD, Mohan S, Baylink D, Linkhart TA. Evidence for a role for insulin-like growth factor binding proteins in glucocorticoid inhibition of normal human osteoblast-like cell proliferation. Eur J Endocrinol. 1996;134:591–601.
Jux C, Leiber K, Hugel U, Blum W, Ohlsson C, Klaus G, Mehls O. Dexamethasone impairs growth hormone (GH)-stimulated growth by suppression of local insulin-like growth factor (IGF)-I production and expression of GH- and IGF-I-receptor in cultured rat chondrocytes. Endocrinology. 1998;139:3296–305.
Knutsen R, Honda Y, Strong DD, Sampath TK, Baylink DJ, Mohan S. Regulation of insulin-like growth factor system components by osteogenic protein-1 in human bone cells. Endocrinology. 1995;136:857–65.
McCarthy TL, Centrella M. Local IGF-I expression and bone formation. Growth Horm IGF Res. 2001;11:213–9.
Mohan S, Baylink DJ. IGF system components and their role in bone metabolism. In: Rosenfeld RG, Roberst C, editors. IGFs in health and diseases. New Jersey: Humana Press; 1999. p. 457–96.
Tremollieres FA, Strong DD, Baylink DJ, Mohan S. Insulin-like growth factor II and transforming growth factor beta 1 regulate insulin-like growth factor I secretion in mouse bone cells. Acta Endocrinol (Copenh). 1991;125:538–46.
Zhang X, Sobue T, Hurley MM. FGF-2 increases colony formation, PTH receptor, and IGF-1 mRNA in mouse marrow stromal cells. Biochem Biophys Res Commun. 2002;290:526–31.
Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82.
Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell. 1993;75:59–72.
Mohan S, Richman C, Guo R, Amaar Y, Donahue LR, Wergedal J, Baylink DJ. Insulin-like growth factor regulates peak bone mineral density in mice by both growth hormone-dependent and -independent mechanisms. Endocrinology. 2003;144:929–36.
Sjogren K, Bohlooly YM, Olsson B, Coschigano K, Tornell J, Mohan S, Isaksson OG, Baumann G, Kopchick J, Ohlsson C. Disproportional skeletal growth and markedly decreased bone mineral content in growth hormone receptor −/− mice. Biochem Biophys Res Commun. 2000;267:603–8.
Mohan S, Baylink DJ. Impaired skeletal growth in mice with haploinsufficiency of IGF-I: genetic evidence that differences in IGF-I expression could contribute to peak bone mineral density differences. J Endocrinol. 2005;185:415–20.
Bikle D, Majumdar S, Laib A, Powell-Braxton L, Rosen C, Beamer W, Nauman E, Leary C, Halloran B. The skeletal structure of insulin-like growth factor I-deficient mice. J Bone Miner Res. 2001;16:2320–9.
Camacho-Hubner C, Woods KA, Clark AJ, Savage MO. Insulin-like growth factor (IGF)-I gene deletion. Rev Endocr Metab Disord. 2002;3:357–61.
Walenkamp MJ, Karperien M, Pereira AM, Hilhorst-Hofstee Y, van Doorn J, Chen JW, Mohan S, Denley A, Forbes B, van Duyvenvoorde HA, et al. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab. 2005;90:2855–64.
Rajaram S, Baylink DJ, Mohan S. Insulin-like growth factor-binding proteins in serum and other biological fluids: regulation and functions. Endocr Rev. 1997;18:801–31.
Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Tornell J, Isaksson OG, Jansson JO, et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci USA. 1999;96:7088–92.
Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA. 1999;96:7324–9.
•• Yakar S, Rosen CJ, Bouxsein ML, Sun H, Mejia W, Kawashima Y, Wu Y, Emerton K, Williams V, Jepsen K, et al. Serum complexes of insulin-like growth factor-1 modulate skeletal integrity and carbohydrate metabolism. FASEB J. 2009;23:709–19. Uses triple knockout mice that lack liver-derived IGF-I, IGFBP-3, and ALS to demonstrate the importance of the circulating IGF regulatory complex in defining skeletal status.
Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR, et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002;110:771–81.
Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A, Efstratiadis A. The hormonal action of IGF1 in postnatal mouse growth. Proc Natl Acad Sci USA. 2008;105:19378–83.
• Nordstrom SM, Tran JL, Sos BC, Wagner KU, Weiss EJ. Liver-derived IGF-I contributes to GH-dependent increases in lean mass and bone mineral density in mice with comparable levels of circulating GH. Mol Endocrinol. 2011;25:1223–30. Reports a significant role for growth hormone-dependent hepatic IGF-I in regulating bone density.
Mohan S, Bautista CM, Wergedal J, Baylink DJ. Isolation of an inhibitory insulin-like growth factor (IGF) binding protein from bone cell-conditioned medium: a potential local regulator of IGF action. Proc Natl Acad Sci USA. 1989;86:8338–42.
Zhao G, Monier-Faugere MC, Langub MC, Geng Z, Nakayama T, Pike JW, Chernausek SD, Rosen CJ, Donahue LR, Malluche HH, et al. Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology. 2000;141:2674–82.
Jiang J, Lichtler AC, Gronowicz GA, Adams DJ, Clark SH, Rosen CJ, Kream BE. Transgenic mice with osteoblast-targeted insulin-like growth factor-I show increased bone remodeling. Bone. 2006;39:494–504.
Devlin RD, Du Z, Buccilli V, Jorgetti V, Canalis E. Transgenic mice overexpressing insulin-like growth factor binding protein-5 display transiently decreased osteoblastic function and osteopenia. Endocrinology. 2002;143:3955–62.
Miyakoshi N, Richman C, Kasukawa Y, Linkhart TA, Baylink DJ, Mohan S. Evidence that IGF-binding protein-5 functions as a growth factor. J Clin Invest. 2001;107:73–81.
Miyakoshi N, Richman C, Qin X, Baylink DJ, Mohan S. Effects of recombinant insulin-like growth factor-binding protein-4 on bone formation parameters in mice. Endocrinology. 1999;140:5719–28.
Qin X, Wergedal JE, Rehage M, Tran K, Newton J, Lam P, Baylink DJ, Mohan S. Pregnancy-associated plasma protein-A increases osteoblast proliferation in vitro and bone formation in vivo. Endocrinology. 2006;147:5653–61.
Richman C, Baylink DJ, Lang K, Dony C, Mohan S. Recombinant human insulin-like growth factor-binding protein-5 stimulates bone formation parameters in vitro and in vivo. Endocrinology. 1999;140:4699–705.
Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, Malluche H, Zhao G, Rosen CJ, Efstratiadis A, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–12.
Govoni KE, Wergedal JE, Florin L, Angel P, Baylink DJ, Mohan S. Conditional deletion of insulin-like growth factor-I in collagen type 1alpha2-expressing cells results in postnatal lethality and a dramatic reduction in bone accretion. Endocrinology. 2007;148:5706–15.
Govoni KE, Lee SK, Chung YS, Behringer RR, Wergedal JE, Baylink DJ, Mohan S. Disruption of insulin-like growth factor (IGF)-I expression in type II{alpha}I collagen expressing cells reduces bone length and width in mice. Physiol Genomics. 2007;30(3):354–62. Epub 2007 May 22.
Wang Y, Cheng Z, Elalieh HZ, Nakamura E, Nguyen MT, Mackem S, Clemens TL, Bikle DD, Chang W. IGF-1R signaling in chondrocytes modulates growth plate development by interacting with the PTHrP/Ihh pathway. J Bone Miner Res. 2011;26:1437–46.
Lean JM, Jagger CJ, Chambers TJ, Chow JW. Increased insulin-like growth factor I mRNA expression in rat osteocytes in response to mechanical stimulation. Am J Physiol. 1995;268:E318–27.
Lean JM, Mackay AG, Chow JW, Chambers TJ. Osteocytic expression of mRNA for c-fos and IGF-I: an immediate early gene response to an osteogenic stimulus. Am J Physiol. 1996;270:E937–45.
Triplett JW, O’Riley R, Tekulve K, Norvell SM, Pavalko FM. Mechanical loading by fluid shear stress enhances IGF-1 receptor signaling in osteoblasts in a PKCzeta-dependent manner. Mol Cell Biomech. 2007;4:13–25.
Xing W, Baylink D, Kesavan C, Hu Y, Kapoor S, Chadwick RB, Mohan S. Global gene expression analysis in the bones reveals involvement of several novel genes and pathways in mediating an anabolic response of mechanical loading in mice. J Cell Biochem. 2005;96:1049–60.
Cheng M, Zaman G, Rawlinson SC, Mohan S, Baylink DJ, Lanyon LE. Mechanical strain stimulates ROS cell proliferation through IGF-II and estrogen through IGF-I. J Bone Miner Res. 1999;14:1742–50.
Cheng MZ, Rawlinson SC, Pitsillides AA, Zaman G, Mohan S, Baylink DJ, Lanyon LE. Human osteoblasts’ proliferative responses to strain and 17beta-estradiol are mediated by the estrogen receptor and the receptor for insulin-like growth factor I. J Bone Miner Res. 2002;17:593–602.
Rawlinson SC, Mohan S, Baylink DJ, Lanyon LE. Exogenous prostacyclin, but not prostaglandin E2, produces similar responses in both G6PD activity and RNA production as mechanical loading, and increases IGF-II release, in adult cancellous bone in culture. Calcif Tissue Int. 1993;53:324–9.
Gross TS, Srinivasan S, Liu CC, Clemens TL, Bain SD. Noninvasive loading of the murine tibia: an in vivo model for the study of mechanotransduction. J Bone Miner Res. 2002;17:493–501.
Sakata T, Halloran BP, Elalieh HZ, Munson SJ, Rudner L, Venton L, Ginzinger D, Rosen CJ, Bikle DD. Skeletal unloading induces resistance to insulin-like growth factor I on bone formation. Bone. 2003;32:669–80.
Sakata T, Wang Y, Halloran BP, Elalieh HZ, Cao J, Bikle DD. Skeletal unloading induces resistance to insulin-like growth factor-I (IGF-I) by inhibiting activation of the IGF-I signaling pathways. J Bone Miner Res. 2004;19:436–46.
Kapur S, Mohan S, Baylink DJ, Lau KH. Fluid shear stress synergizes with insulin-like growth factor-I (IGF-I) on osteoblast proliferation through integrin-dependent activation of IGF-I mitogenic signaling pathway. J Biol Chem. 2005;280:20163–70.
Lau KH, Kapur S, Kesavan C, Baylink DJ. Up-regulation of the Wnt, estrogen receptor, insulin-like growth factor-I, and bone morphogenetic protein pathways in C57BL/6J osteoblasts as opposed to C3H/HeJ osteoblasts in part contributes to the differential anabolic response to fluid shear. J Biol Chem. 2006;281:9576–88.
•• Long RK, Nishida S, Kubota T, Wang Y, Sakata T, Elalieh HZ, Halloran BP, Bikle DD. Skeletal unloading-induced insulin-like growth factor 1 (IGF-1) nonresponsiveness is not shared by platelet-derived growth factor: the selective role of integrins in IGF-1 signaling. J Bone Miner Res. 2011;26:2948–58. Reports that skeletal unloading causes bone loss that is associated with impaired IGF-I but not PDGF signaling.
•• Kesavan C, Wergedal JE, Lau KH, Mohan S. Conditional disruption of IGF-I gene in type 1alpha collagen-expressing cells shows an essential role of IGF-I in skeletal anabolic response to loading. Am J Physiol Endocrinol Metab 301:E1191–7. Reports that locally produced IGF-I is critically involved in mediating bone formation response to mechanical strain in mice.
Acknowledgments
Financial support was received from funding agencies in the United States (NIH grant AR048139 and VA merit review grant).
Disclosures
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mohan, S., Kesavan, C. Role of Insulin-like Growth Factor-1 in the Regulation of Skeletal Growth. Curr Osteoporos Rep 10, 178–186 (2012). https://doi.org/10.1007/s11914-012-0100-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-012-0100-9