
Abstract
Purpose of Review
In this review, we highlight the pre-clinical development, recent clinical studies, and future directions of larotrectinib in patients with NTRK fusion-positive tumors.
Recent Findings
The tropomyosin receptor kinase family, TrkA, TrkB, and TrkC, transmit extracellular signals via a variety of intracellular pathways to promote normal neuronal development. TrkA, B, and C are encoded by NTRK1, 2, and 3, respectively. NTRK chromosomal alterations, most commonly gene fusions, have been identified as driver mutations in a broad range of malignancies. Small molecule tyrosine kinase inhibitors of Trk, including larotrectinib, have shown broad clinical activity across multiple tumor types with NTRK fusion events.
Summary
Although the prevalence of NTRK alterations is low, the exceptional activity of larotrectinib makes NTRK alterations an important predictive biomarker to screen for in any cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The tropomyosin receptor kinases TRKA, TRKB, and TRKC belong to a family of receptor tyrosine kinases (RTK) that are critically involved in neural development [1, 2]. TRKs are composed of an extracellular domain responsible for ligand binding, a membrane-spanning domain, and an intracellular ATP-binding domain involved in the modulation of downstream signaling pathways [3]. TRKA, TRKB, and TRKC bind several neurotrophins: nerve growth factor, brain-derived growth factor, and neurotrophin 3/4. Binding of these neurotrophins leads to receptor dimerization and autophosphorylation [4]. Activation of TRK triggers a cascade of signaling pathways including the MAPK, PI3K, and PLCγ pathways which are important for cell proliferation, differentiation, survival, and angiogenesis [5,6,7,8,9].
The genes neurotrophic tyrosine receptor kinase 1, 2, and 3 (NTRK1, NTRK2, NTRK3) encode TRKA, TRKB, and TRKC, respectively [4]. Genomic aberrations involving NTRK that have been shown to drive tumorigenesis include gene fusions, single-nucleotide substitutions, in-frame deletions, and alternative splicing [10,11,12,13]. Of these, intra-chromosomal and inter-chromosomal rearrangements are the most common genetic events driving tumorigenesis [14]. Aberrant gene products form as a result of chromosomal rearrangement of the 3′ end of the NTRK proto-oncogene to the 5′ end of an unrelated gene [13]. The resultant protein product causes inappropriate activation of the TRK kinase domain and leads to unopposed activation of several cell proliferation and growth pathways involved in oncogenesis [15]. Over 20 partner genes have been reported to occur with variable frequency including ETV6, PAN3, and TPM3 [10].
TRK fusions, while rare, have been reported to occur in a diverse set of cancer types [16]. There are an estimated 1500 to 5000 cases of TRK fusion-positive cancers in the USA annually [17]. Common cancers that have been associated with NTRK fusion events include lung adenocarcinoma, sarcoma, acute myeloid leukemia, and colorectal cancer although the overall incidence in each of these tumor types remains quite low [18]. NTRK fusions have recently been reported to occur in patients with neuroendocrine tumors, a tumor type which has not previously been associated with a driver mutation (Table 1) [16, 19•]. Several rare tumors types that have been associated with pathognomonic ETV6-NTRK3 gene fusions include secretory breast carcinoma, mammary analog secretory carcinoma, congenital fibrosarcomas, and congenital mesoblastic nephroma [15, 20, 21•, 22, 23].
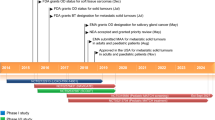
With increased appreciation for the prevalence as well as oncogenic potential of NTRK fusion events across tumor types, there has been considerable interest in developing effective tyrosine kinase inhibitors (TKI) to abrogate aberrant TRK signaling. Larotrectinib (LOXO-101) is a highly selective pan-TRK inhibitor that has shown considerable efficacy in patients with an NTRK fusion-positive cancer. In this review, we describe the pre-clinical and clinical data leading to the FDA granting breakthrough therapy designation to larotrectinib for the treatment of TRK fusion-positive tumors regardless of histology.
Pre-clinical Studies
Larotrectinib binds and competitively inhibits the ATP-binding site of TRKA, TRKB, and TRKC. Larotrectinib interferes with autophosphorylation of the kinase domain of TRK and thereby diminishes downstream signaling [24, 25]. Maximum plasma concentration of larotrectinib is achieved 30–60 min after dosing. Ninety-eight percent inhibition of TRKA, TRKB, and TRKC is achieved at all dose levels [26, 27]. IC50 levels are reported in the low nanomolar range [10, 26, 27].
Larotrectinib demonstrated dose-dependent inhibition of cell proliferation in various cell lines with associated NTRK gene fusions. Specifically, larotrectinib showed considerable in vitro activity in three cancer cell lines including CUTO-3.29 derived from a patient with lung adenocarcinoma harboring MPRIP-NTRK gene fusion, KM12 derived from a patient with colorectal adenocarcinoma harboring TPM3-NTRK1, and MO-91 derived from a patient with acute myeloid leukemia harboring ETV6-NTRK3 [24]. Of note, larotrectinib has no in vitro activity in cell lines lacking NTRK gene fusions [25]. In vivo dose-dependent activity was further demonstrated in athymic nude mice injected with KM12 cells [24]. Given the promising in vitro and in vivo results, clinical studies were undertaken to evaluate the safety and efficacy of larotrectinib in patients with tumors harboring NTRK fusions.
Clinical Studies
The first published report of the activity of larotrectinib in a human subject occurred in a 41-year-old woman diagnosed with a soft tissue undifferentiated sarcoma with metastatic pulmonary involvement [24]. Comprehensive genomic profiling (CGP) was performed utilizing techniques previously reported [28, 29]. Gene fusion of exons 1–2 of lamin A/C (LMNA) and exons 11–17 of NTRK1 was detected. The absence of other putative oncogenic mutations on CGP suggested the LMNA-NTRK1 fusion gene was driving tumorigenesis. After several lines of conventional local and systemic therapies, the patient was enrolled on the LOXO-101 clinical study (NCT02122913). Four months after initiating larotrectinib, the patient experienced a complete response.
Several case reports further suggested the considerable clinical activity of larotrectinib in patients with NTRK fusion-positive tumors. A 14-year-old female from Bangladesh with advanced secretory breast carcinoma was found to have an ETV6-NTRK3 gene fusion. The patient experienced a near complete response after 2 months of therapy [30]. Additional benefit was seen in a pediatric patient with relapsed infantile fibrosarcoma associated with ETV6-NTRK3 gene fusion who experienced a partial response to larotrectinib [31]. The activity of larotrectinib in CNS was first suggested by a patient with non-small cell lung cancer with metastases to the brain with TPR-NTRK1 fusion. The patient experienced a CNS response after treatment with larotrectinib [32]. Recently, a 3-year-old girl with a refractory high-grade glioma was found to have ETV6-NTRK3 fusion. Treatment with larotrectinib resulted in near complete response sustained at 9 months of therapy [33]. While most responses have been reported in patients with metastatic disease, the potential role of larotrectinib as pre-operative therapy was recently demonstrated in four pediatric patients with locally advanced TRK fusion-positive sarcomas. Four of five patients with refractory disease were able to proceed with definitive surgical resection after a median of 6 cycles of larotrectinib [34].
Based on promising initial clinical data, larotrectinib received breakthrough therapy designation in July 2016 by the Food and Drug Administration (FDA) for adult and pediatric patients with advanced solid tumors positive for NTRK fusions. Of considerable interest, it was the first therapy to receive this designation for a tissue-agnostic indication.
The integrated safety and efficacy results of 55 patients enrolled in three early-phase clinical trials were reported in patients with NTRK-positive solid tumors (Table 2) [35••]. Eight patients were from an adult phase 1 trial, 12 patients were from a phase 1/2 pediatric trial (SCOUT), and 35 patients (at least 12 years of age) were from a phase 2 basket trial (NAVIGATE). All patients had locally advanced or metastatic solid tumors. Larotrectinib was administered at a dose of 100 mg by mouth twice daily. The median age of patients enrolled was 45 years; 77% of patients were 15 years or older. Seventeen diverse tumor types were represented with the most common histologies being salivary gland carcinoma (22%), infantile fibrosarcoma (13%), thyroid carcinoma (7%), colon cancer (7%), lung cancer (7%), and melanoma (7%). TRK fusions status was determined by local CLIA-accredited laboratories and involved NTRK1 in 45%, NTRK2 in 2%, and NTRK3 in 53% of patients respectively.
The objective response rate (ORR) by RECIST v1.1 (independent review) was 75% (95% confidence interval, 61 to 85). The complete response rate was 13% and the partial response rate was 62%. Two patients with infantile fibrosarcoma were able to proceed with curative limb-sparing resection. Reponses occurred irrespective of tumor type, age, or TRK fusion subtype. The median time to response was 1.8 months (range 0.9 to 6.4). The median duration of response was 8.3 months. The median progression-free survival was not reached at 9.9 months of follow-up. The responses observed were durable with 55% of patients remaining free of progression at 1 year of follow-up. Responses were ongoing at the time of data cutoff in 71% of patients.
Larotrectinib was found to be well tolerated with only 15% of patients requiring dose reduction. The most common adverse events grade 3 or higher in severity related to larotrectinib were anemia (11%), increased alanine aminotransferase or asparate aminotransferase level (7%), weight gain (7%), and decreased neutrophil count (7%). There were no grade 4 or 5 events felt to be related to the study agent. Of note, no patient with a response required drug discontinuation related to an adverse event. Taken together, larotrectinib is safely administered at therapeutic dosing with few dose-limiting toxicities.
Future Directions
As with other tyrosine kinase inhibitors, mechanisms of acquired resistance represent not only a therapeutic challenge but also an opportunity with the development of second-generation agents which overcome resistance mechanisms. Among patients initially experiencing an objective response to larotrectinib, 23% later developed progressive disease. Of these patients, 90% had an identifiable secondary resistance mutation in either NTRK1 (G595R, G667S, F589L) or NTRK3 (G623R, G696A) [35••]. Loxo-195 is a selective TRK tyrosine kinase inhibitor which overcomes resistance to first-generation TKIs [36]. This agent is effective against resistance mutations which impact both the solvent front and xDFG domains of the TRK enzyme and has shown preliminary efficacy in two patients with acquired resistance to larotrectinib [36]. Currently, NCT03215511 is a multi-center, phase 1/2 clinical trial designed to evaluate the safety and efficacy of LOXO-195 in patients with NTRK-rearranged tumors treated with a prior TRK inhibitor. In addition to LOXO-195, two additional novel agents, TPX-0005 and ONO-5390556, have demonstrated pre-clinical activity in tumors with acquired resistance to first-generation TRK inhibitors [37, 38]. The development of second- and third-generation TRK inhibitors may well lead to patients experiencing prolonged tumor responses analogous to progress made with EGFR-, ALK-, and ROS1-directed therapies.
Furthermore, the preferred diagnostic modality to detect rare TRK fusion events has not been defined. Several approaches to detect NTRK fusion events are currently available including fluorescence in situ hybridization (FISH), immunohistochemistry, reverse-transcriptase polymerase chain reaction (RT-PCR), and next-generation sequencing of either DNA or RNA. DNA-based next-generation sequencing (NGS) allows for detection of NTRK fusion events along with multiple other genomic alterations. This approach is particularly attractive in tumor types where numerous genomic alterations are of therapeutic interest. Nonetheless, caution must be exercised even with DNA-based NGS approaches. This is a particular consideration with regard to NTRK fusions involving NTRK2 and NTRK3 by virtue of large intronic regions which may confound testing results [39]. Targeted RNA sequencing may have advantages over DNA-based NGS techniques in that unknown upstream partners may be more readily detectable. One large series of patients with lung adenocarcinomas revealed gene fusions involving NTRK not previously found with DNA-based NGS [40••].
Conclusions
NTRK fusion-positive tumors have increasingly been seen as a rare but important genomic driver of tumorigenesis across a diverse landscape of tumor types. Inhibitors of TRK have demonstrated significant activity in pre-clinical models. Larotrectinib exhibits broad clinical activity across multiple tumor histologies harboring NTRK fusions, regardless of organ of origin. Identification of resistance mutations to first-generation TRK inhibitors has resulted in the development of additional TRK inhibitors that overcome these mutations. As these newer TRK inhibitors reach the clinic, genomic surveillance at progression will enable sequenced TRK inhibitor therapy further impacting the natural history of NTRK fusion-positive tumors.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Arévalo JC, Wu SH. Neurotrophin signaling: many exciting surprises! Cell Mol Life Sci. 2006;63(13):1523–37.
Nakagawara A. Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett. 2001;169(2):107–14.
Klein R, Jing SQ, Nanduri V, O’Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65(1):189–97.
Kheder ES, Hong DS. Emerging targeted therapy for tumors with NTRK fusion proteins. Clin Cancer Res. 2018;24(23):5807–14.
Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991;350(6314):158–60.
Kawamura K, Kawamura N, Fukuda J, Kumagai J, Hsueh AJW, Tanaka T. Regulation of preimplantation embryo development by brain-derived neurotrophic factor. Dev Biol. 2007;311(1):147–58.
Kawamura K, Kawamura N, Sato W, Fukuda J, Kumagai J, Tanaka T. Brain-derived neurotrophic factor promotes implantation and subsequent placental development by stimulating trophoblast cell growth and survival. Endocrinology. 2009;150(8):3774–82.
Kawamura K, Kawamura N, Kumazawa Y, Kumagai J, Fujimoto T, Tanaka T. Brain-derived neurotrophic factor/tyrosine kinase B signaling regulates human trophoblast growth in an in vivo animal model of ectopic pregnancy. Endocrinology. 2011;152(3):1090–100.
Loeb DM, Stephens RM, Copeland T, Kaplan DR, Greene LA. A Trk nerve growth factor (NGF) receptor point mutation affecting interaction with phospholipase C-gamma 1 abolishes NGF-promoted peripherin induction but not neurite outgrowth. J Biol Chem. 1994;269(12):8901–10.
Khotskaya YB, Holla VR, Farago AF, Mills Shaw KR, Meric-Bernstam F, Hong DS. Targeting TRK family proteins in cancer. Pharmacol Ther. 2017;173:58–66.
Miranda C, Mazzoni M, Sensi M, Pierotti MA, Greco A. Functional characterization of NTRK1 mutations identified in melanoma. Genes Chromosom Cancer. 2014;53(10):875–80.
Tacconelli A, Farina AR, Cappabianca L, Desantis G, Tessitore A, Vetuschi A, et al. TrkA alternative splicing: a regulated tumor-promoting switch in human neuroblastoma. Cancer Cell. 2004;6(4):347–60.
Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5(1):25–34.
Greco A, Miranda C, Pierotti MA. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol Cell Endocrinol. 2010;321(1):44–9.
Rubin JB, Segal RA. Growth, survival and migration: the Trk to cancer. Cancer Treat Res. 2003;115:1–18.
Sigal DS, Bhangoo MS, Hermel JA, Pavlick DC, Frampton G, Miller VA, et al. Comprehensive genomic profiling identified novel NTRK fusions in neuroendocrine tumors. Oncotarget. 2018;9(88):35809–12.
Hyman DM, Laetsch TW, Kummar S, DuBois SG, Farago AF, Pappo AS, et al. The efficacy of larotrectinib (LOXO-101), a selective tropomyosin receptor kinase (TRK) inhibitor, in adult and pediatric TRK fusion cancers. J Clin Oncol. 2017;35(18_suppl):LBA2501.
Chen Y, Chi P. Basket trial of TRK inhibitors demonstrates efficacy in TRK fusion-positive cancers. J Hematol Oncol 2018 7 [cited 2018 Aug 28];11. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5992878/
• Sigal D, Tartar M, Xavier M, Bao F, Foley P, Luo D, et al. Activity of entrectinib in a patient with the first reported NTRK fusion in neuroendocrine cancer. J Natl Compr Cancer Netw. 2017;15(11):1317–22 This paper highlights the utility of NTRK inhibition in a patient with metastatic neuroendocrine tumor, a tumor type not previously known to exhibit targetable driver mutations.
Liu D, Offin M, Harnicar S, Li BT, Drilon A. Entrectinib: an orally available, selective tyrosine kinase inhibitor for the treatment of NTRK, ROS1, and ALK fusion-positive solid tumors. Ther Clin Risk Manag. 2018;14:1247–52.
• Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2(5):367–76 This report characterizes the molecular pathogenesis of a secretory breast carcinoma, a rare histologic subtype associated with a TRK fusion event.
Watanabe N, Kobayashi H, Hirama T, Kikuta A, Koizumi S, Tsuru T, et al. Cryptic t(12;15)(p13;q26) producing the ETV6-NTRK3 fusion gene and no loss of IGF2 imprinting in congenital mesoblastic nephroma with trisomy 11: fluorescence in situ hybridization and IGF2 allelic expression analysis. Cancer Genet Cytogenet. 2002;136(1):10–6.
Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18(2):184–7.
Doebele RC, Davis LE, Vaishnavi A, Le AT, Estrada-Bernal A, Keysar S, et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. 2015 Oct;5(10):1049–57.
Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19(11):1469–72.
Hong DS, Brose MS, Doebele RC, Shaw AT, Dowlati A, Bauer TM, et al. Abstract PR13: clinical safety and activity from a phase 1 study of LOXO-101, a selective TRKA/B/C inhibitor, in solid-tumor patients with NTRK gene fusions. Mol Cancer Ther. 2015;14(12 Supplement 2):PR13.
Burris HA, Shaw AT, Bauer TM, Farago AF, Doebele RC, Smith S, et al. Abstract 4529: pharmacokinetics (PK) of LOXO-101 during the first-in-human phase I study in patients with advanced solid tumors: interim update. Cancer Res. 2015;75(15 Supplement):4529.
Bhangoo MS, Boasberg P, Mehta P, Elvin JA, Ali SM, Wu W, et al. Tumor mutational burden guides therapy in a treatment refractory POLE-mutant uterine carcinosarcoma. Oncologist. 2018;23(5):518–23.
Bhangoo MS, Costantini C, Clifford BT, Chung JH, Schrock AB, Ali SM, et al. Biallelic deletion of PALB2 occurs across multiple tumor types and suggests responsiveness to poly (ADP-ribose) polymerase inhibition. JCO Precis Oncol. 2017;1:1–7.
Shukla N, Roberts SS, Baki MO, Mushtaq Q, Goss PE, Park BH, et al. Successful targeted therapy of refractory pediatric ETV6-NTRK3 fusion-positive secretory breast carcinoma. JCO Precis Oncol. 2017. https://doi.org/10.1200/PO.17.00034.
Nagasubramanian R, Wei J, Gordon P, Rastatter JC, Cox MC, Pappo A. Infantile fibrosarcoma with NTRK3-ETV6 fusion successfully treated with the tropomyosin-related kinase inhibitor LOXO-101. Pediatr Blood Cancer. 2016;63(8):1468–70.
Hong DS, Dowlati A, Burris HA III, Lee JJ, Brose MS, Farago AF, et al. Clinical safety and activity from a phase 1 study of LOXO-101, a selective TRKA/B/C inhibitor, in solid-tumor patients with NTRK gene fusions. Eur J Cancer. 2017;72:S148.
Ziegler DS, Wong M, Mayoh C, Kumar A, Tsoli M, Mould E, et al. Brief report: potent clinical and radiological response to larotrectinib in TRK fusion-driven high-grade glioma. Br J Cancer. 2018;119(6):693–6.
DuBois SG, Laetsch TW, Federman N, Turpin BK, Albert CM, Nagasubramanian R, et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer. 2018;124(21):4241–7.
•• Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731–9 Landmark study reporting the safety and efficacy results of three early-phase clinical trials utilizing larotrectinib across multiple tumor types (pediatric and adults).
Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017;7(9):963–72.
Zhai D, Deng W, Huang J, Rogers E, Cui JJ. Abstract 3161: TPX-0005, an ALK/ROS1/TRK inhibitor, overcomes multiple resistance mechanisms by targeting SRC/FAK signaling. Cancer Res. 2017;77(13 Supplement):3161.
Kozaki R, Yoshizawa T, Tsukamoto K, Kato H, Kawabata K. Abstract 2954A: a potent and selective TRK inhibitor ONO-5390556, shows potent antitumor activity against both TRK-rearranged cancers and the resistant mutants. Cancer Res. 2016 Jul 15;76(14 Supplement):2954A.
Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15(12):731–47.
•• Benayed R, Offin MD, Mullaney KA, Sukhadia P, Rios KM, Desmeules P, et al. Comprehensive detection of targetable fusions in lung adenocarcinomas by complementary targeted DNAseq and RNAseq assays. J Clin Oncol. 2018;36(15_suppl):12076 This study emphasizes the sensitivity of RNA sequencing techniques which may identify TRK fusions not seen with conventional DNA sequencing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Munveer S. Bhangoo declares that he has no conflict of interest.
Darren Sigal has a patent issued on a method of treating neuroendocrine tumors.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Evolving Therapies
Rights and permissions
About this article

Cite this article
Bhangoo, M.S., Sigal, D. TRK Inhibitors: Clinical Development of Larotrectinib. Curr Oncol Rep 21, 14 (2019). https://doi.org/10.1007/s11912-019-0761-y
Published:
DOI: https://doi.org/10.1007/s11912-019-0761-y