
Abstract
The increasing incidence of thyroid cancer is associated with a higher number of advanced disease characterized by the loss of cancer differentiation and metastatic spread. The knowledge of the molecular pathways involved in the pathogenesis of thyroid cancer has made possible the development of new therapeutic drugs able to blockade the oncogenic kinases (RET/PTC) or signaling kinases (vascular endothelial growth factor receptor [VEGFR]) involved in cellular growth and proliferation. Some clinical trials have been conducted showing the ability of targeted therapies able to inhibit RET (sorafenib, imatinib, vandetanib) in stabilizing the course of the disease. The aim of the introduction of these targeted therapies is to extend life duration assuring a good quality of life; however, further studies are needed to reach these goals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thyroid cancer is the seventh most common cause of any new malignancy in the US for women (25,480 cases in 2007, 3.8% of new cases), while causing only about 1% of new cases in men (8070 cases in 2007; incidence rank: 14th) [1–3]. The yearly incidence of thyroid cancer has been growing from 7800 cases in 1974 to 33,550 cases in 2007. During the same period, the percentage of cancer deaths per year, relative to the number of new cases, has decreased from 15% to 5% [1–3]. Using a large data base on thyroid cancer it has been found [4] that 79% of cases were papillary (PTC), 13% follicular, 3% Hurthle cell, 3% medullary (MTC), and 2% anaplastic. PTC presents most commonly between 30 and 50 years of age, with a female/male ratio of 2–3:1. The risk of PTC is increased in subjects exposed to radiations [5, 6].
The increasing incidence of thyroid cancer is associated with a higher number of advanced disease characterized by the loss of cancer differentiation and metastatic spread [7], causing high morbidity, but not necessarily death.
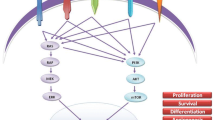
The knowledge of the molecular pathways involved in the pathogenesis of thyroid cancer has made possible the development of new therapeutic drugs able to blockade the oncogenic kinases (BRAF V600E, RET/PTC) or signaling kinases (vascular endothelial growth factor receptor [VEGFR], platelet-derived growth factor receptors [PDGFR]) involved in cellular growth and proliferation [8].
RET
In the thyroid gland, RET is highly expressed in parafollicular C-cells but not in follicular cells, where it can be activated by chromosomal rearrangement known as RET/PTC rearrangement [9]. The RET gene is located on chromosome 10q11.2 [10]. RET encodes a transmembrane receptor with an extracellular portion containing four calcium-dependent cell-adhesion domains needed to interact with ligands [11]. The extracellular portion of the receptor also contains multiple glycosylation sites and a cysteine-rich region necessary for receptor dimerization [9]. The intracellular domain of the RET receptor contains two tyrosine kinase regions that activate intracellular signal transduction pathways. RET activation triggers autophosphorylation of tyrosine residues that serve as docking sites for adaptor proteins, which coordinate cellular signal transduction pathways (eg, mitogen-activated protein kinase, phosphatidylinositol 3-kinase, etc.) which are important in the regulation of cell growth [9].
Papillary Thyroid Cancer
In PTC RET can be activated by chromosomal rearrangement known as RET/PTC rearrangement [9].
In RET/PTC, the 3′ portion of the RET gene is fused to the 5′ portion of various unrelated genes. At least 13 types of RET/PTC have been reported to date, all formed by the RET fusion to different partners [9]. The two most common rearrangement types, RET/PTC1 and RET/PTC3, account for the vast majority of all rearrangements found in papillary carcinomas. RET/PTC1 is formed by fusion with the H4 (D10S170) gene, and RET/PTC3 by fusion with the NCOA4 (ELE1) gene [9].
In RET/PTC rearrangements, fusion with protein partners possessing protein-protein interaction motifs provides RET/PTC kinases with dimerizing interfaces, which results in ligand-independent autophosphorylation. The RET intracellular domain contains at least 12 autophosphorylation sites, 11 of which are maintained in RET/PTC proteins [12]. Tyrosine 905 (Y905) is a binding site for Grb7/10 adaptors [13], Y1015 for phospholipase Cγ [14], and Y981 for c-Src [15]. Tyrosine 1062 is the binding site for several proteins, including the Shc proteins, insulin receptor substrate–1/2 (IRS-1/2), FGFR substrate 2 (FRS2), downstream of kinase 1/4/5 (DOK1/4/5), and Enigma, which, in turn, lead to the activation of many signaling pathways [14]. Binding to Shc and FRS2 mediates recruitment of Grb2-SOS complexes, which thus leads to GTP exchange on RAS and RAS/ERK stimulation [16]. RET-PTC has recently been shown to be able to activate also another important oncogenic pathway in thyroid carcinoma, the PI3K pathway [16].
RET/PTC is tumorigenic in thyroid follicular cells, as it transforms thyroid cells in culture and gives rise to thyroid carcinomas in transgenic mice [17].
Several studies suggest that the oncogenic effects of RET/PTC require signaling along the MAPK pathway and the presence of the functional BRAF kinase. Indeed, BRAF silencing in cultured thyroid cells reverses the RET/PTC-induced effects [18].
In PTC, RET/PTC rearrangements are found in 30–40%, RAS mutations in about 10%, and BRAF mutations in approximately 40–50%, with no overlap among these mutations, whereas a higher prevalence of BRAF mutations (up to 70%) has been observed in dedifferentiated papillary thyroid carcinoma (DePTC) [19, 20]. RET/PTC rearrangements are correlated to radiation exposure and are found in pediatric PTC [21]. Twelve variants of RET/PTC have been described [22].
Papillary carcinomas with RET/PTC rearrangements typically present at younger age and have a high rate of lymph node metastases, classic papillary histology, and possibly more favorable prognosis, particularly those harboring RET/PTC1. In tumors arising after radiation exposure, RET/PTC1 was found to be associated with classic papillary histology, whereas RET/PTC3 type was more common in the solid variants [23, 24].
Correlation between RET/PTC rearrangement and prognosis in human papillary carcinomas remains unclear. Some evidence suggests that the RET/PTC1 rearrangement type is associated with more favorable behavior of papillary carcinomas [9].
Medullary Thyroid Carcinoma
MTC originates from the calcitonin (CT)-producing neuroendocrine cells of the thyroid gland and accounts for 5% of all thyroid malignancies [25, 26].
MTC is usually a slow-growing tumour, and patients with metastatic disease have 10-year overall survival rates of 40–50% [27]. Most cases of MTC are sporadic and present with metastatic disease at diagnosis; approximately 20% of MTC cases occur as a component of multiple endocrine neoplasia syndrome type 2 (MEN2). Surgery remains the only curative modality available [28]; however, there are no effective therapeutic options available for distant metastatic disease because the tumor does not respond to radiotherapy or chemotherapy.
Activating mutations of the tyrosine kinase receptor (TKR) RET have been reported in nearly all hereditary cases of MTC and in 30–50% of sporadic tumor cases [29]. Thus, RET has become an important therapeutic target for MTC.
MEN2A is the most common (50%) form of hereditary MTC. There are three variants of MEN2A. The first is associated with Hirschsprung disease [30], the second is associated with cutaneous lichen amyloidosis [31], while the third variant, familial MTC (FMTC) [32], is characterized by MTC but no other manifestations of MEN2. MEN2A is characterized by bilateral, multicentric MTC in more than 90% of index patients (carrying germline RET mutations in codon 634; i.e., those presenting with MTC at diagnosis in the absence of screening), pheochromocytomas in 50%, and primary hyperparathyroidism in 10% to 20%.
MEN2B is less common than MEN2A (approximately 20% of cases of MEN2) [33]. MEN2B is characterized by MTC in 100% of carriers, pheochromocytomas in 50%, mucosal ganglioneuromas in more than 90%, and a marphanoid habitus in nearly all. Early identification of MTC in MEN2B is important because metastases have been described during the first year of life.
The importance of RET activation in MTC has been discussed extensively in some prior reviews [9, 12, 34].
Although dysregulation of the RET pathway remains the most important event in the development of MTC [35], a recent study demonstrated interactions between the RET and another TKR, the epidermal growth factor receptor (EGFR). The EGFR contributed to RET kinase activation, signalling, and growth stimulation [36]. This reinforces the concept that RET may cooperate with diverse transduction pathways and signals to promote MTC tumorigenesis and may contribute to stimulation of other as yet unknown cellular signalling cascades [34].
Current Therapies for Treatment of Thyroid Cancers
Tyrosine Kinase Receptor Inhibitors
Tyrosine kinase inhibitors are small organic compounds that affect tyrosine kinase–dependent oncogenic pathways, competing with the ATP-binding site of the catalytic domain of the tyrosine kinase [37]. Occupation of this site inhibits autophosphorylation and activation of the tyrosine kinase and prevents further activation of intracellular signalling pathways. TKIs can be specific to one or several homologous tyrosine kinases. Thus, a single TKI may target multiple tyrosine kinases [38].
Several small-molecule tyrosine kinase inhibitors directed toward RET kinase have been tested in in vitro, preclinical, and clinical studies (Tables 1 and 2). ZD6474, an orally active low-molecular-weight receptor tyrosine kinase inhibitor, is a potent inhibitor of the vascular endothelial growth factor receptor 2 (VEGFR-2) and effectively blocks RET tyrosine kinase [39]. ZD6474 has been shown to block phosphorylation and signaling from RET/PTC3 and RET carrying the most common MEN2A and MEN2B mutations in vitro, to induce growth arrest of human papillary carcinoma cell lines carrying RET/PTC1, and to prevent tumor growth in nude mice after injection of RET/PTC3-transformed fibroblasts or RET mutation-positive medullary carcinoma cells [40, 41].
Since antitumor activity of ZD6474 is likely due to a combination of its anti-RET activity and antiangiogenic activity mediated by blocking VEGFR, it will be of importance to find whether the extent of the therapeutic response to ZD6474 depends on the presence of RET mutation and its type. In preclinical studies, ZD6474 has been shown to inhibit most of the mutated variants of RET, except for the V804L and V804M mutations [42]. V804 in the RET protein corresponds to the gate-keeper residues of ABL, PDGFR, c-KIT, and EGFR kinases, and mutations at these residues are known to confer resistance to various inhibitors [43]. These results suggest that RET V804L and V804M mutations in medullary carcinomas may mediate primary resistance to ZD6474 [42].
Various classes of small TKIs have shown inhibition of RET activity in preclinical studies, including pyrazolopyrimidine inhibitors PP1 and PP2, 2-indolinone derivative RPI-1, and indolocarbazole derivatives CEP-701 and -751 [44–47]. PP1 and PP2 have been tested in preclinical studies and found to be effective in therapeutic concentrations in blocking RET/PTC signaling in vivo and abolishing its tumorigenic effects in experimental animals. A multikinase inhibitor SU12248 (sunitinib) has been shown to effectively inhibit signaling from RET/PTC kinase in the experimental models and has been progressed to phase II clinical trial in radioiodine-refractory, unresectable differentiated thyroid cancer and medullary thyroid cancer.
Trials of Different Tyrosine Kinase Inhibitors with RET-Inhibiting Activity for the Treatment of Thyroid Cancer
Since 2005, a wide variety of multitargeted kinase inhibitors have entered clinical trials for patients with advanced or progressing metastatic thyroid cancers yielding higher response rates than cytotoxic chemotherapy, also if responses have been observed in only few patients [48, 49]. Most of these agents have a common property of inhibiting VEGFR, with a potent anti-angiogenetic role, because of the structural similarity between RET and VEGFR kinases; ie, sorafenib has RAF-RET, and VEGFR-inhibiting activity; imatinib has RET, and VEGFR-2 inhibiting activity; vandetanib inhibits VEGFR-2, EGFR, and RET. Other agents act on different pathways, for example: axitinib has VEGFR-, C-KIT-, and platelet-derived growth factor receptors (PDGFR)-inhibiting activity; pazopanib is an inhibitor of VEGFR and PDGFR; and sunitinib inhibits E7080 (a multi-kinase inhibitor) and VEGFR.
Several phase II trials of different tyrosine kinase inhibitors against RET were conducted and others are ongoing (Table 1).
There have been different phase II trials of sorafenib (BAY 43–9006, 400 mg twice daily). The first trial of sorafenib (400 mg twice daily) was conducted on 30 differentiated thyroid carcinoma (DTC) patients, and a partial response was reported in 7 patients and stable disease in 16 patients [50••]. The second trial reported a partial response in 6 patients and stable disease for 6 months or more in 23 of the 41 patients with PTC, but therapy was ineffective in the 11 patients with FTC or poorly differentiated DTC and in the 4 patients with ATC [51••]. In the third trial, which was conducted on 32 DTC patients, a partial response was reported in 8 patients and stable disease in 11 patients [52••]. Greater efficacy of sorafenib was seen in PTC, especially PTC with a BRAF mutation, than in poorly differentiated DTC, and it had no effect on iodine uptake. A phase III trial comparing progression-free survival is under way, in which sorafenib and placebo are being administered to patients with radioiodine-resistant metastatic DTC.
A further phase II trial of sorafenib in patients with advanced MTC (16 sporadic, or 25 hereditary) has been conducted. Among 16 patients with sporadic MTC, one achieved partial response (6.3%), 14 had stable disease (87.5%), and one was non-evaluable. Median progression-free survival was 17.9 months. The treatment in MTC hereditary patients was prematurely terminated because of slow accrual [53••].
Recently, Capdevila et al. tested sorafenib in patients with advanced thyroid carcinoma [54••]. Thirty patients were enrolled and, among 27 evaluable patients, they observed partial responses in nine (30%) and stable disease in 13 patients. With a median follow-up of 11 months, the median progression-free survival was 11.6 months. Interestingly, six out of 12 (50%) patients with MTC obtained a partial response, indicating that the drug might be effective in MTC, although the small number of patients requires further prospective studies.
On the basis of these promising results, a multicenter, double-blind randomized phase III study evaluating the efficacy and safety of sorafenib in locally advanced/metastatic RAI-refractory differentiated thyroid cancer [55••, 56••] and an international, randomized, double-blinded phase III efficacy study of XL184 in subjects with unresectable, locally advanced, or metastatic MTC [57••], both compared to placebo, are ongoing.
Recently, Verbeek et al. [58•] compared the effect of four TKIs (axitinib, sunitinib, vandetanib, and XL184) on cell proliferation, RET expression, and autophosphorylation and extracellular signal-regulated kinases (ERK) activation in cell lines expressing MEN2A, MEN2B mutations, and a RET/PTC rearrangement, showing a most potent effect of XL184 (an inhibitor of VEGFR-1 and -2, C-MET [59], RET, C-KIT, FLT3, and Tie-2) in MEN2A and PTC and a more effective action of vandetanib in MEN2B in vitro.
A phase I dose-escalation study of oral XL184 (cabozantinib) was conducted in 37 patients with MTC. Ten (29%) of 35 patients with MTC with measurable disease had a confirmed partial response. Additionally, 15 (41%) of 37 patients with MTC had stable disease for at least 6 months, resulting in stable disease for 6 months or longer or confirmed partial response in 68% of patients with MTC [60••].
The TKI imatinib (STI571) was tested in patients with metastatic MTC yielding no objective responses but stable disease in a minority of patients [61], while motesanib, in a mouse model of MTC, inhibited tumor xenograft growth reducing directly VEGFR-2 and RET expression [62•]. In a metastatic murine model of MTC, it has been recently tested a novel RET inhibitor, withaferin A, that was responsible for tumor regression and growth delay [63•].
A phase II study assessed the efficacy of vandetanib (300 mg/day), a selective inhibitor of VEGFR-2, epidermal growth factor receptor (EGFR), and RET, in patients with advanced hereditary MTC. Thirty patients were enrolled: 20% of patients had a partial response, while an additional 53% of patients experienced a stable disease at 24 weeks. A reduction in calcitonin and carcinoembryonic antigen levels was also reported [64••]. Another study evaluated vandetanib in 19 patients with advanced hereditary MTC, yielding a partial response in 3 patients, and stable disease lasting 24 weeks or longer in further 10 patients with decreasing levels of calcitonin and carcinoembryonic antigen levels (a sustained 50% or greater decrease from baseline in 3 of 19 patients and 5% in 1 of 19 patients, respectively) [65••].
In April 2011, the US Food and Drug Administration (FDA) approved vandetanib (AstraZeneca) for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease [66••]. It is the first drug ever approved for the treatment of this rare form of thyroid cancer. The approval of vandetanib is based on the results of the phase III ZETA study, which randomized 331 patients with unresectable locally advanced or metastatic MTC to vandetanib 300 mg (n = 231) or placebo (n = 100). In the study, patients treated with vandetanib had a median progression-free survival of at least 22.6 months, compared with 16.4 months for patients randomized to placebo (hazard ratio [HR], 0.35; 95% confidence interval [CI], 0.24–0.53; P < 0.0001). Serious adverse events reported during the study resulted in five deaths in patients treated with vandetanib. Causes of death included breathing complications, heart failure, and sepsis. Vandetanib has been shown to affect the electrical activity of the heart, which in some cases can cause irregular heartbeats that can be life-threatening. The prescribing information for vandetanib includes a box warning about treatment-related QT prolongation, Torsades de pointes, and sudden death. The most common adverse drug reactions (> 20%) seen in the ZETA trial with vandetanib were diarrhea (57%), rash (53%), acne (35%), nausea (33%), hypertension (33%), headache (26%), fatigue (24%), decreased appetite (21%), and abdominal pain (21%), according to the company.
Targeted Therapies Limits
Tumor cells often devise strategies to bypass the effects of antineoplastic agents and selection of therapy-resistant clones is frequently the reason for treatment failure. A lack of response can occur, for instance, because target inhibition raises the activity of compensatory signal pathways, which, in turn, rescue tumor cell growth. However, the possibility of testing the sensitivity of primary DePTC cells from each subject to different TKIs could increase the effectiveness of the treatment. In fact, in vitro chemosensitivity tests are able to predict in vivo effectiveness in 60% of cases [67]; while, it is well known that a negative chemosensitivity test in vitro is associated with a 90% of ineffectiveness of the treatment in vivo [68], allowing the administration of inactive chemotherapeutics to these patients to be avoided [67]. It has been recently demonstrated that it is possible to test the antineoplastic activity of different compounds (antiblastics or peroxisome proliferator-activated receptor [PPAR]-γ agonists) in primary anaplastic thyroid cancer cells obtained from each patient [69•, 70•]. Furthermore, it has been shown that primary cells can be obtained directly from fine needle aspiration samples of thyroid cancer, and that the results of in vitro chemosensitivity tests are quite similar to those obtained from surgical biopsies [71•]. Interestingly, more recently, we have first shown an antitumoral effect of two (CLM3 and CLM29) new multi-targeted kinases inhibitors (CLM3 and CLM29 inhibit several targets, including RET, EGFR, and VEGFR and have an antiangiogenic effect) not on continuous cell lines, that are quite different from the tumor of the patients themselves, but directly on primary DePTC of patients refractory to the radioiodine therapy [72••], opening the way to the possibility of personalizing the kinase inhibitors therapy in each patient.
Targeted Therapies Side Effects
Numerous side effects of the multi-targeted kinase inhibitors were reported; in different trials, several patients required a dose reduction to improve tolerability. The most common side effects involved cardiovascular system (hypertension, cardiomyopathy, stroke) and skin (rashes, foot and hand syndrome) [48]. Some cases of TSH elevation were also observed, especially during motesanib therapy [73].
Several endocrine effects were observed in patients with vandetanib, a multikinase inhibitor (targeting also RET) in 39 patients with progressive thyroid cancer. During vandetanib treatment, several changes were observed: 1) serum 25(OH) vitamin D level decreased and serum PTH and 1,25(OH)(2) vitamin D levels increased, suggesting a decreased intestinal absorption of vitamin D or lack of sun exposure as a result of photosensitization; 2) L-T(4) doses were to maintain serum TSH within the normal range; 3) in male patients, total testosterone and free testosterone levels increased; and 4) serum inhibin B decreased and stimulated FSH increased, suggesting a Sertoli cells insufficiency [74].
Conclusions
The most encouraging results, in patients with advanced PTC unresponsive to radioiodine therapy and MTC, were obtained with the targeted kinase inhibitors with an intrinsic activity against VEGFR and cross activity against RET kinases, such as sorafenib, imatinib, and vandetanib.
Until now, no consensus guidelines or standard criteria for patients enrolling were established definitively. Thereby, the effects on survival are unclear, because of lack of complete responses and because of the discrepancies between the radiographic tumor responses and the effective improvement of survival.
The aim of the introduction of these targeted therapies is to extend life duration assuring a good quality of life. To reach these goals, we need to have further data on toxicities of single agent and of combination of more drugs, to identify specific biomarkers able to predict the treatment efficacy, the clinical outcome, and to guarantee a tailored dosage of the drugs. Moreover, the possibility to test in vitro (in primary thyroid cancer cells) these novel drugs may help to improve further the personalization of the treatment.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA. 2006;295(18):2164–7.
Ries LAG, et al (eds). SEER cancer statistics review, 1975–2001, National Cancer Institute, Bethesda, MD, 2004. Available at: seer.cancer.gov/csr/1975–2001.
Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66.
Hundahl SA, Fleming ID, Fremgen AM, et al. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985–1995 [see commetns]. Cancer. 1998;83(12):2638–48.
Antonelli A, Miccoli P, Derzhitski VE, et al. Epidemiologic and clinical evaluation of thyroid cancer in children from the Gomel region (Belarus). World J Surg. 1996;20(7):867–71.
Antonelli A, Fallahi P, Grosso M, et al. Lobectomy versus total thyroidectomy in children with post-Chernobyl thyroid cancer: a 15 year follow-up. Endocrine. 2011 Jun 23.
Wartofsky L. Increasing world incidence of thyroid cancer: increased detection or higher radiation exposure? Hormones (Athens). 2010;9(2):103–8.
Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol. 2011;7(10):569–80.
de Groot JW, Links TP, Plukker JT, et al. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocr Rev. 2006;5:535–60.
Pasini B, Hofstra RM, Yin L, et al. The physical map of the human RET proto-oncogene. Oncogene. 1995;11(9):1737–43.
Anders J, Kjar S, Ibanez CF. Molecular modeling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin-like domains and calcium-binding site. J Biol Chem. 2001;276(38):35808–17.
Plaza-Menacho I, Burzynski GM, De Groot JW, et al. Current concepts in RET-related genetics, signaling and therapeutics. Trends Genet. 2006;22(11):627–36.
Pandey A, Duan H, Di Fiore PP, et al. The Ret receptor protein tyrosine kinase associates with the SH2-containing adapter protein Grb10. J Biol Chem. 1995;270(37):21461–3.
Salvatore D, Barone MV, Salvatore G, et al. Tyrosines 1015 and 1062 are in vivo autophosphorylation sites in ret and ret-derived oncoproteins. J Clin Endocrinol Metab. 2000;85(10):3898–907.
Kato M, Takeda K, Kawamoto Y, et al. Repair by Src kinase of function-impaired RET with multiple endocrine neoplasia type 2A mutation with substitutions of tyrosines in the COOH-terminal kinase domain for phenylalanine. Cancer Res. 2002;62(8):2414–22.
Santarpia L, Myers JN, Sherman SI, et al. Genetic alterations in the RAS/RAF/mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways in the follicular variant of papillary thyroid carcinoma. Cancer. 2010;116(12):2974–8.
Santoro M, Chiappetta G, Cerrato A, et al. Development of thyroid papillary carcinomas secondary to tissue-specific expression of the RET/PTC1 oncogene in transgenic mice. Oncogene. 1996;12(8):1821–6.
Mitsutake N, Miyagishi M, Mitsutake S, et al. BRAF mediates RET/PTC-induced mitogen-activated protein kinase activation in thyroid cells: functional support for requirement of the RET/PTC-RAS-BRAF pathway in papillary thyroid carcinogenesis. Endocrinology. 2006;147(2):1014–9.
Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353(2):172–87.
Antonelli A, Fallahi P, Ferrari SM, et al. Dedifferentiated thyroid cancer: a therapeutic challenge. Biomed Pharmacother. 2008;62(8):559–63.
Fenton CL, Lukes Y, Nicholson D, et al. The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. J Clin Endocrinol Metab. 2000;85(3):1170–5.
Santoro M, Dathan NA, Berlingieri MT, et al. Molecular characterization of RET/PTC3; a novel rearranged version of the RETproto-oncogene in a human thyroid papillary carcinoma. Oncogene. 1994;9(2):509–16.
Powell Jr DJ, Russell J, Nibu K, et al. The RET/PTC3 oncogene: metastatic solid-type papillary carcinomas in murine thyroids. Cancer Res. 1998;58(23):5523–8.
Nikiforov YE, Rowland JM, Bove KE, et al. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 1997;57(9):1690–4.
Antonelli A, Ferrari SM, Fallahi P, et al. Medullary thyroid cancer: new targeted molecular therapies. Recent Pat Endocr Metab Immune Drug Discov. 2010;4(1):10–4. 5.
Ball DW, Baylin SB, De Butros AC. Medullary thyroid carcinoma. In: BravermanLE UtigerRD, editor. Werner and Ingbar’s the thyroid. 8th ed. Philadelphia: Lippincott Williams and Wilkins; 2000. p. 930–43.
Kebebew E, Clark OH. Medullary thyroid cancer. Curr Treat Options Oncol. 2000;1(14):359–67.
Weber T, Shilling T, Buchler MW. Thyroid carcinoma. Curr Opin Oncol. 2006;18(1):30–5.
Drosten M, Pützer BM. Mechanisms of Disease: cancer targeting and the impact of oncogenic RET for medullary thyroid carcinoma therapy. Nat Clin Pract Oncol. 2006;3(10):564–74.
Verdy M, Weber AM, Roy CC, et al. Hirschsprung’s disease in a family with multiple endocrine neoplasia type 2. J Pediatric Gastroenterol Nutr. 1982;1(14):603–7.
Gagel RF, Levy ML, Donovan DT, et al. Multiple endocrine neoplasia type 2a associated with cutaneous lichen amyloidosis. Ann Intern Med. 1989;111(10):802–6.
Farndon JR, Leight GS, Dilley WG, et al. Familial medullary thyroid carcinoma without associated endocrinopathies: a distinct clinical entity. Br J Surg. 1986;73(4):278–81.
Williams ED, Pollock DJ. Multiple mucosal neuromata with endocrine tumours: a syndrome allied to von Recklinghausen’s disease. J Pathol Bacteriol. 1966;91(1):71–80.
Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005;16(4–5):441–67.
Castellone MD, Santoro M. Dysregulated RET signaling in thyroid cancer. Endocrinol Metab Clin North Am. 2008;37(2):363–74.
Croyle M, Akeno N, Knauf JA, et al. RET/PTC-induced cell growth is mediated in part by epidermal growth factor receptor (EGFR) activation: evidence for molecular and functional interactions between RET and EGFR. Cancer Res. 2008;68(11):4183–91.
Lorusso PM, Eder JP. Therapeutic potential of novel selective-spectrum kinase inhibitors in oncology. Expert Opin Investig Drugs. 2008;17(7):1013–28.
Antonelli A, Ferri C, Ferrari SM, et al. New targeted molecular therapies for dedifferentiated thyroid cancer. J Oncol. 2010;2010:921682.
Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62(24):7284–90.
Vieira JM, Santos SC, Espadinha C, et al. Expression of vascular endothelial growth factor (VEGF) and its receptors in thyroid carcinomas of follicular origin: a potential autocrine loop. Eur J Endocrinol. 2005;153(5):701–9.
Elliott DD, Sherman SI, Busaidy NL, et al. Growth factor receptors expression in anaplastic thyroid carcinoma: potential markers for therapeutic stratification. Hum Pathol. 2008;39(1):15–20.
McGregor LM, McCune BK, Graff JR, et al. Roles of trk family neurotrophin receptors in medullary thyroid carcinoma development and progression. Proc Natl Acad Sci USA. 1999;96(13):4540–5.
Mitsiades CS, Kotoula V, Poulaki V, et al. Epidermal growth factor receptor as a therapeutic target in human thyroid carcinoma: mutational and functional analysis. J Clin Endocrinol Metab. 2006;91(9):3662–6.
Cuccuru G, Lanzi C, Cassinelli G, et al. Cellular effects and antitumor activity of RET inhibitor RPI-1 on MEN2A-associated medullary thyroid carcinoma. J Natl Cancer Inst. 2004;96(13):1006–14.
Strock CJ, Park JI, Rosen M, et al. CEP-701 and CEP-751 inhibit constitutively activated RET tyrosine kinase activity and block medullary thyroid carcinoma cell growth. Cancer Res. 2003;63(17):5559–63.
Carniti C, Perego C, Mondellini P, et al. PP1 inhibitor induces degradation of RETMEN2A and RETMEN2B oncoproteins through proteosomal targeting. Cancer Res. 2003;63(9):2234–43.
Carlomagno F, Vitagliano D, Guida T, et al. Efficient inhibition of RET/papillary thyroid carcinoma oncogenic kinases by 4-amino-5-(4-chloro-phenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2). J Clin Endocrinol Metab. 2003;88(4):1897–902.
Schlumberger M. Kinase inhibitors for refractory thyroid cancers. Lancet Oncol. 2010;11(10):912–3.
Sherman SI. Targeted therapy of thyroid cancer. Biochem Pharmacol. 2010;80(5):592–601.
•• Gupta-Abramson V, Troxel AB, Nellore A, et al. Phase II trial of sorafenib in advanced thyroid cancer. J Clin Oncol. 2008;26(29):4714–9. Phase II clinical trial of sorafenib in thyroid cancer.
•• Kloos RT, Ringel MD, Knopp MV, et al. Phase II trial of sorafenib in metastatic thyroid cancer. J Clin Oncol. 2009;27(10):1675–84. Phase II clinical trial of sorafenib in thyroid cancer.
•• Hoftijzer H, Heemstra KA, Morreau H, et al. Beneficial effects of sorafenib on tumor progression, but not on radioiodine uptake, in patients with differentiated thyroid carcinoma. Eur J Endocrinol. 2009;161(6):923–31. Phase II clinical trial of sorafenib in thyroid cancer.
•• Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010;28(14):2323–30. Phase II clinical trial of sorafenib in thyroid cancer.
•• Capdevila J, Iglesias L, Halperin I, et al. Sorafenib in patients (pts) with advanced thyroid carcinoma (TC): a compassionate use program. J Clin Oncol 28(Suppl. 15). 2010 (2010 ASCO Annual Meeting, Abstract 5590). Phase II clinical trial of sorafenib in thyroid cancer.
•• Nexavar® Versus Placebo in Locally Advanced/Metastatic RAI-Refractory Differentiated Thyroid Cancer (NCT00984282) http://clinicaltrials.gov/ct2/show/NCT00984282?term=NCT00984282&rank=1. Phase III clinical trial of sorafenib in thyroid cancer.
•• Brose MS, Nutting CM, Sherman SI, et al. Rationale and design of decision: a double-blind, randomized, placebo-controlled phase III trial evaluating the efficacy and safety of sorafenib in patients with locally advanced or metastatic radioactive iodine (RAI)-refractory, differentiated thyroid cancer. BMC Cancer. 2011;11:349. Phase III clinical trial of sorafenib in thyroid cancer.
•• Efficacy of XL184 (Cabozantinib) in Advanced Medullary Thyroid Cancer http://clinicaltrials.gov/ct2/show/NCT00704730?term=phase+III%2C+thyroid+cancer&rank=4 (NCT00704730). Phase III clinical trial of XL184 in thyroid cancer.
• Verbeek HH, Alves MM, de Groot JW, et al. The Effects of Four Different Tyrosine Kinase Inhibitors on Medullary and Papillary Thyroid Cancers Cells. J Clin Endocrinol Metab. 2011; 96(6):E991–5. In vitro study of tyrosine kinase inhibitors in medullary thyroid cancer cells.
Cui JJ. Inhibitors targeting hepatocyte growth factor receptor and their potential therapeutic applications. Expert Opin Ther Pat. 2007;17(9):1035–45.
•• Kurzrock R, Sherman SI, Ball DW, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29(19):2660–6. Phase II clinical trial of cabozantinib in thyroid cancer.
de Groot JW, Zonnenberg BA, van Ufford-Mannesse PQ, et al. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2007;92(9):3466–9.
• Coxon A, Bready J, Estrada J, et al. Antintumor Activity of Motesanib in a Medullary Thyroid Cancer Model. J Endocrinol Invest. 2011 Mar 21. In vitro study of tyrosine kinase inhibitors in medullary thyroid cancer cells.
• Samady AK, Mukerji R, Shah A, et al. A novel RET inhibitor with potent efficacy against medullary thyroid cancer in vivo. Surgery. 2010;148(6):1228–36. In vitro study of tyrosine kinase inhibitors in medullary thyroid cancer cells.
•• Wells SA Jr, Gosnell JE, Gagel RF, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28(5):767–72. Phase II clinical trial of vandetanib in thyroid cancer.
•• Robinson BG, Paz-Ares L, Krebs A, et al. Vandetanib (100 mg) in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Endocrinol Metab. 2010;95(6):2664–71. Phase II clinical trial of vandetanib in thyroid cancer.
•• Commander H, Whiteside G, Perry C. Vandetanib: first global approval. Drugs. 2011;71(10):1355–65. Phase II clinical trial of vandetanib in thyroid cancer.
Blumenthal RD, Goldenberg DM. Methods and goals for the use of in vitro and in vivo chemosensitivity testing. Mol Biotechnol. 2007;35(2):185–97.
Sawyers CL. Disabling Abl-perspectives on Abl kinase regulation and cancer therapeutics. Cancer Cell. 2002;1(1):13–5.
• Antonelli A, Ferrari SM, Fallahi P, et al. Thiazolidinediones and antiblastics in primary human anaplastic thyroid cancer cells. Clin Endocrinol (Oxf). 2009;70(6):946–53. In vitro first study evaluating sensitivity to chemotherapeutics and thiazolidinediones in cells obtained from anaplastic thyroid cancer.
• Antonelli A, Ferrari SM, Fallahi P, et al. Evaluation of the sensitivity to chemotherapeutics or thiazolidinediones of primary anaplastic thyroid cancer cells obtained by fine-needle aspiration. Eur J Endocrinol. 2008;159(3):283–91. In vitro first study evaluating sensitivity to chemotherapeutics and thiazolidinediones in cells obtained by fine-needle aspiration.
• Antonelli A, Ferrari SM, Fallahi P, et al. Primary cell cultures from anaplastic thyroid cancer obtained by fine-needle aspiration used for chemosensitivity tests. Clin Endocrinol (Oxf). 2008;69(1):148–52. In vitro first study evaluating sensitivity to chemotherapeutics in cells obtained by fine-needle aspiration.
•• Antonelli A, Bocci G, La Motta C, et al. Novel pyrazolopyrimidine derivatives as tyrosine kinase inhibitors with antitumoral activity in vitro and in vivo in papillary dedifferentiated thyroid cancer. J Clin Endocrinol Metab. 2011:96(2):E288–96. First study showing the effect of tyrosine kinase inhibitors in primary cells obtained from DePTC.
Schlumberger MJ, Elisei R, Bastholt L, et al. Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. J Clin Oncol. 2009;27(23):3794–801.
Brassard M, Neraud B, Trabado S, et al. Endocrine effects of the tyrosine kinase inhibitor vandetanib in patients treated for thyroid cancer. J Clin Endocrinol Metab. 2011;96(9):2741–9.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Antonelli, A., Fallahi, P., Ferrari, S.M. et al. RET TKI: Potential Role in Thyroid Cancers. Curr Oncol Rep 14, 97–104 (2012). https://doi.org/10.1007/s11912-012-0217-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-012-0217-0