
Abstract
Medullary thyroid carcinoma (MTC) is a hyperplasia of thyroid C-cells, accounting for 5–10% of all thyroid cancers. MTCs may appear as sporadic or hereditary forms, and several molecules and signaling pathways have been found to function defectively in MTC cells. Tyrosine kinases are the most well-studied molecules that have abnormal function in these tumor cells. Due to their limited response, chemotherapeutic agents and radiation therapy are not effective in treating patients with advanced metastatic MTC. In the past decade, significant attention has been given to the utilization of multikinase inhibitors as targeted therapeutic agents for treating MTC patients, with the most promising results arising from the study of tyrosine kinase inhibitors, which generally bind to the ATP binding sites of these kinases. Two drugs—vandetanib and cabozantinib—are approved for the treatment of aggressive advanced MTC; however, the potential for toxicities and adverse effects of these agents on patient quality of life need to be considered against any therapeutic gain. According to recent data, it appears that inhibition of only one receptor or molecule in a pathway is not as effective as simultaneous inhibition of different pathways, indicating the need to use combination therapy. The main purpose of this review is to describe the clinical characteristics, molecular mechanisms, and current molecular and targeted therapeutic strategies active in clinical trials for advanced MTC treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A better understanding of impaired molecular mechanisms involved in medullary thyroid cancer (MTC) will lead to the identification of more specific and targeted drugs for the treatment of MTC. |
Cabozantinib and vandetanib are approved for the management of unresectable, advanced tumors. |
Patient quality of life and side effects of drugs must be weighed against the potential benefit of these agents. |
1 Introduction
Medullary thyroid carcinoma (MTC) is a neuroendocrine tumor arising from parafollicular C-cells of the thyroid gland, and accounts for 5–10% of all thyroid cancers, with a 1–2% incidence in nodular thyroid disorders [1, 2]. More than 100 years ago, Jaquet described a thyroid tumor with amyloid [3], while, in 1959, Hazard et al. elucidated histological features of this tumor and proposed the term ‘medullary thyroid carcinoma’ [4]. These features include spindle-shaped, round or polygonal cells segregated by fibrous stroma, a pattern that is a common characteristic of endocrine tumors. The cytoplasm of cells in these tumors is eosinophilic and granular, and nuclei are predominantly uniform round to oval [5]. Furthermore, animal studies showed that MTCs were derived from the parafollicular cells of the thyroid [6]. In 1967, Bussolati and Pearse [7] showed that parafollicular cells are responsible for the production and secretion of calcitonin, and subsequent studies confirmed the presence of calcitonin in tumor extracts and the serum of MTC patients [8, 9]. The discovery of specific mutations and the abnormal expression of certain proteins, such as the tyrosine kinase receptors, as well as some key factors in the signaling pathways, could help to understand the biology, clinical characteristics, treatment, and prognosis of MTCs. Recent scientific developments have helped to better understand the management of progressive and advanced MTC [10]. To date, surgery is the main and only curative treatment for MTC. Chemotherapeutic agents such as dacarbazine, 5-fluorouracil, and doxorubicin are not efficient, showing partial responses (PRs) in the range of 10–20%, and are of short duration. Over the past few years, a number of multikinase inhibitors have been evaluated for treating advanced MTC tumors [11, 12], and a number of molecular mechanisms involved in MTC development have been demonstrated, but there is room for further improvement. Tyrosine kinases are the most investigated molecules in pathways that have abnormal function in MTC tumors, for example, the receptor tyrosine kinases (RTKs) family have an aberrant function in tumor cells [10, 13]. This review will discuss the clinical charateristics and molecular mechanisms that underlie MTC, and provide a review of the molecular and targeted therapeutics currently undergoing clinical trials.
2 Clinical Characteristics
MTCs may be either sporadic or hereditary. Hereditary tumors are associated with multiple endocrine neoplasia (MEN) 2 syndrome: MEN2A, MEN2B and familial MTC (FMTC), which is considered a subtype of MEN2A [14]. Moreover, sporadic forms account for 75% of cases, while MEN2-associated cases account for the remainder [15, 16]. Table 1 summarizes the clinical characteristics of medullary thyroid cancer subtypes.
Sporadic MTC is primarily a tumor that occurs around the ages 40–60 years, with a female to male ratio of 1.3:1. Kebebew et al. reported that approximately 75% of patients develop a thyroid mass, while 15% presented with symptoms of dysphagia, dyspnea, or hoarseness [17]. In this study, nearly 10% of patients had systemic symptoms. Cushing’s syndrome may also occur in approximately 0.6% of patients with MTC, and survival following diagnosis of the syndrome is poor [18]. In the MEN2 syndromes, penetrance of MTCs is very high, and it is estimated that more than 90% of carriers will develop thyroid tumors [19].
MEN2A accounts for almost more than 34% of all hereditary MTCs [20]. The most common abnormalities that can occur in MEN2A patients are pheochromocytomas (PHEOs; in 40–60% of cases) and hyperparathyroidism (HPTH) (in 10–30% of cases). It has been reported that in nearly 10% of patients with MEN2A, PHEO may precede the appearance of the thyroid tumors. In this syndrome, PHEOs are commonly bilateral and multicentric, and hyperplasia of the adrenal medulla may be the precursor of PHEO [21, 22]. The presence of cutaneous lichen amyloidosis, pruritic plaques located on the upper back, is also reported in some kindreds with MEN2A or FMTC [23], and some families with MEN-2A may also have Hirschsprung’s disease [24]. Patients with MEN2A usually develop thyroid tumors around early adulthood; however, calcitonin screening studies and molecular diagnostic testing has seen the age at diagnosis become progressively younger [19, 23].
MEN2B is another subtype of MEN2 syndrome, with a distinctive phenotype, it often presents in infancy and accounts for approximately 6.8% of the hereditary forms of MTC [20]. In contrast to MEN2A, patients with MEN2B have no observable parathyroid abnormalities, however 40–60% of cases have PHEOs. Furthermore, affected patients also have neuromas of the tongue and ganglioneuromatosis of the intestine, with or without marfanoid signs. It has been reported that MTCs in this syndrome have an early onset and are highly aggressive, metastasizing to regional lymph nodes and beyond. Leboulleux et al. showed that the stage at diagnosis of MTC is the major prognostic factor in patients with MEN2B [19, 25, 26].
FMTC, a subtype of MEN2A, is a form of medullary carcinoma that has the least aggressive features [27], and is the most common form of hereditary MTC (57.6%) [20]. Patients with FMTC typically develop no other clinical manifestations, such as HPTH or PHEO, and the age at which FMTC presents is the same as those with nonfamilial MTCs. Interestingly, molecular analysis revealed that a remarkable proportion of patients presenting with apparent sporadic MTCs will prove to have FMTC [28].
3 Pathways of Molecular Pathogenesis
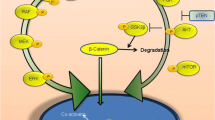
There are several molecules and signaling pathways that are functioning aberrantly in MTC cells. In this section we describe some of these molecules and how they can participate in MTC development (Fig. 1).

Different molecules and pathways that are involved in MTC pathogenesis. MTC medullary thyroid carcinoma, RET rearranged during transfection, VEGFR vascular endothelial growth factor receptor, EGFR epidermal growth factor receptor, HGFR hepatocyte growth factor receptor, FGFR fibroblast growth factor receptor
3.1 Rearranged During Transfection Receptor
The rearranged during transfection (RET) proto-oncogene is located on chromosome 10q11.2 and is composed of 21 exons and has a size of approximately 55,000 base pairs [29]. RET encodes an RTK which has an extracellular component that contains cadherin-like domains and multiple glycosylation sites. Moreover, this portion of RET also has a cysteine-rich region, which is necessary for maintaining the tertiary structure of the protein and receptor dimerization. The intracellular domain has two tyrosine kinase regions that activate the intracellular cascade of signal transduction. To activate RET, a number of ligands, such as glial cell line-derived neurotrophic factor (GDNF), neurturin, artemin, or persephin, must bind to a cell membrane coreceptor, glycosylphosphatidylinositol-anchored GDNF family receptor (GFR)-α [30,31,32]. Interaction of this complex with RET results in its dimerization, followed by autophosphorylation of the tyrosine residues on the intracellular domains. These residues serve as docking sites for adaptor proteins, which are necessary to trigger pathways, such as the mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase pathway (PI3K/AKT), and c-Jun N-terminal protein kinases (JNK) [33]. The RET oncogene was discovered by Takahashi et al. [34]. Thereafter, it was found that virtually all patients with MEN2A, MEN2B, and FMTC have RET germline mutations, and nearly 65% of patients with sporadic MTC may have somatic mutations in RET [35,36,37,38,39]. RET mutations typically result in constitutive activation of receptor, and are therefore called gain-of-function mutations [40]. Some studies have suggested a prognostic role for RET. The somatic RET codon M918T mutation in sporadic MTC seems to anticipate an aggressive clinical course and a poor prognosis [41]. This mutation results in unfortunate consequences for the patients, they have the highest probability of persistent disease and also a lower survival rate at long-term follow-up [42]. Moura et al. also reported that among sporadic MTC, cases with RET mutations in exons 15 and 16 are associated with the worst prognosis. Cases with other RET mutations have the most indolent course, and those with no RET mutations have an intermediate risk [43].
It has been reported that 20–30% of patients with papillary thyroid cancer (PTC) have activating chromosomal translocations in RET [44]. These translocations also appear in patients with lung adenocarcinoma and chronic myelomonocytic leukemia [45, 46]. Moreover, some RET mutations are known as RET janus mutations, these are a small group of mutations in exon 10: C620, C618, C611 and C609. Janus mutations act simultaneously, as both a gain-of-function and loss-of-function mutation. RET janus mutations are known to cause co-occurrence of Hirschsprung’s disease and MEN2A [47]. To date, more than 100 mutations, duplications, insertions, or deletions in RET have been identified in patients with hereditary MTC. The data for these germline mutations are available online at http://www.arup.utah.edu/database/MEN2/MEN2_welcome.php [48].
3.2 Vascular Endothelial Growth Factor (VEGF) and VEGF Receptors
Abnormal angiogenesis, an important feature of tumors, is essential for tumor progression and metastasis. Interaction of vascular endothelial growth factors (VEGFs) and VEGF receptors (VEGFRs) regulate several biological processes such as angiogenesis, vascular permeability and endothelial cell proliferation, migration, and survival of tissue. To date, four types of VEGFs (A, B, C, and D) and three types of VEGFRs (1, 2, and 3) have been discovered. Binding of VEGF to its receptor leads to activation of the MAPK and PI3K/AKT signaling pathways, which in turn leads to angiogenesis and vasculogenesis [49, 50]. It is noted that different VEGFs can bind to the different VEGFRs: VEGF-A binds to both VEGFR-1 and VEGFR-2; VEGF-B binds to VEGFR-1; and VEGF-C and VEGF-D are specific for VEGFR-3 [50]. It is believed that VEGFR-2 is the major mediator of MTC angiogenesis [51]. Moreover, it has been shown that VEGF-A, VEGFR-1, VEGFR-2, and VEGFR-3 are overexpressed in MTC, and overexpression of VEGFR-2 was associated with an increased rate of metastases [52,53,54].
3.3 Epidermal Growth Factor Receptor
The epidermal growth factor receptor (EGFR), also called HER-1/ErbB-1, is an RTK that regulates cell growth and apoptosis, and is frequently overexpressed in MTC. EGFR is a member of the ErbB family, which includes EGFR (ErbB-1), HER2/c-neu (ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4). The binding of ligands such as epidermal growth factor (EGF), transforming growth factor-α (TGFα), and neuregulins to these receptors leads to the dimerization and activation of EGFR, which can trigger the MAPK and PI3K pathways [55,56,57]. Several mechanisms have been described for oncogenic activation of EGFR: overexpression of ligand or EGFR, activating mutations, deficiency in transactivation, or inactivation mechanisms [58]. Croyle et al. revealed the ability of RET to form a complex with EGFR, and reported that ligand-induced activation of EGFR can result in phosphorylation of a kinase-dead RET, an effect that is entirely blocked by PKI166. In addition, EFGR kinase inhibitors impair growth of MTC cell lines carrying the C634W mutation and expressing a constitutively active RET protein [59]. Rodríguez-Antona et al. showed that EGFR polysomies and the expression of this receptor in tumors with the most aggressive RET mutations (M918T, A883F) were significantly decreased compared with the other RET mutations. These data suggest that the most aggressive RET mutations are less dependent on EGFR activation; however, researchers found that EGFR was significantly overexpressed in metastases compared with primary tumors [52].
3.4 Hepatocyte Growth Factor Receptor
Hepatocyte growth factor receptor (HGFR), also known as MET or c-Met proto-oncogene, is a receptor that, in humans, is encoded by the MET gene [60]. Abnormal activation of HGFR induces tumor growth, angiogenesis, invasion, and metastasis [61]. A normal human thyroid has abundant levels of cMET, which are important in normal thyroid function and thyroid neoplasia [62]. Activating mutations and constitutive overexpression of cMET have been found in many tumor cells [63,64,65,66,67,68,69,70], while it has also been reported that silencing of MET could impair invasive growth in vitro and in vivo [61]. Missense mutations have been found in the c-MET gene in some MTC samples. Furthermore, some studies have shown coexpression of MET and hepatocyte growth factor (HGF) in a subset of MTC, in which autocrine/paracrine circuits may be active. These investigations have suggested that there is no correlation between HGF/MET expression and clinicopathological parameters, except for the multicentric feature of HGF/MET-positive MTC [71, 72]. There is crosstalk between MET and RET at transcriptional and signaling levels, therefore the inhibition of RET and MET can block cell proliferation and invasiveness in some papillary thyroid carcinoma cell lines [73]. Furthermore, activation of RAS and RET oncogenes results in highly expressed c-MET in human thyroid epithelial cells [74].
3.5 Fibroblast Growth Factor Receptor-4
Fibroblast growth factor receptor (FGFR)-4 is a member of the FGFR family [75]. Overexpression of FGFR-4 in aggressive tumors and the most rapidly proliferative cell lines, indicates that it may promote the progression of these tumors [76]. Preclinical experiments have shown that the inhibition of growth factor receptors such as FGFR-4 reduces the growth of MTC cells [77]; thus, this receptor may be a therapeutic target in patients with progressive MTC.
3.6 Rat Sarcoma
The rat sarcoma (RAS) gene encodes three distinct but highly homologous 21 kDa proteins, including H-RAS, K-RAS, and N-RAS. These small guanosine triphosphatases (GTPases) have central regulatory roles in several signaling pathways, such as the MAPK and PI3K/Akt pathways, which control a wide range of effects, including cell proliferation, differentiation, and apoptosis [78]. Activating point mutations in RAS genes, which have been extensively described in several tumor types, constitutively activate multiple downstream effectors [79, 80]. Recently, HRAS and KRAS mutations were reported in MTC. RAS mutations in sporadic MTC have a prevalence of between 0 and 43.3% and occur mostly in tumors with normal RET, and rarely in those with mutated RET, suggesting that activation of these proto-oncogenes represents alternative genetic events in sporadic MTC tumorigenesis [81]. Some studies have found that these mutations in RAS are not associated with germline or somatic RET mutations, suggesting that these mutations are mutually exclusive [82, 83]. MTC patients with RAS mutations have a lower risk for aggressive tumor than those with RET mutations in exons 15 and 16 [81].
4 The Role of Next-Generation Sequencing in MTC Mutation Profiling
Next-generation sequencing (NGS) is a method used to identify molecular alterations likely causative of the tumoral transformation [84]. Marina et al. conducted a study to explore the use of targeted NGS to simultaneous test for multiple mutations in thyroid cancer [86]. They designed a custom panel (ThyroSeq) to target 12 cancer genes with 284 mutational hot spots. Eleven of 15 MTCs (73%) were positive for mutations and the most common mutation was RET p.M918T, followed by RAS mutations. In addition, the most common type of RAS mutation was HRAS p.Q61K, followed by KRAS p.G12R and HRAS p.G13R [85]. Polymerase chain reaction (PCR) amplification and sequencing analysis of the three mutational hotspots (codons 12, 13, and 61) of the H-, K-, and N-RAS genes, and of the mutational hotspot (codon 600) and exon 11 of the BRAF gene in 65 sporadic MTCs, of which 40 were RET positive and 25 were RET negative, showed that 68.0% of RET-negative sporadic MTCs had somatic RAS mutations, respectively; however, only 2.5% of RET-positive sporadic MTCs had an RAS mutation [86]. Another study was performed in a total of 12 cases of sporadic (9/12) and hereditary MTCs (3/12), the results of which showed that two of three hereditary MTCs harbored RET/C634R mutations, while the other one had two RET mutations (L790F and S649L). All the sporadic MTCs had RET/M918T mutation, with the exception of one case with HRAS mutation [87]. Using multigene Ion AmpliSeq NGS technology, Simbolo et al. analyzed 50 cancer-related genes in 20 sporadic MTCs. Of 20 MTC samples, 13 had RET mutations, three cases had HRAS mutations, and one case had KRAS mutations. Of 13 RET-mutated MTCs, one case harbored a germline mutation in STK11. The three remaining MTCs (15%) were wild-type for all 50 cancer-related genes [88]. The role of RET fusion as an oncogenic driver in MTC has also been investigated in a patient who died from aggressive sporadic MTC, using NGS that included an intronic capture strategy. The data obtained in this study revealed an in-frame fusion transcript joining MYH13 exon 35 with RET exon 12 [89]. Although RET gene fusions are the most well known rearrangements found in MTCs, recent data shows the other prominent gene fusions that occur in this type of thyroid cancer. Ji et al. conducted a study to investigate the genetic profile of 84 sporadic MTC samples and 36 paired normal thyroid tissues. Of the 84 MTC tissue samples, the study found the MTC harbored mutations as follows: 47 RET mutations, 14 HRAS mutations, 11 serine/threonine kinase 11 mutations, 6 KIT mutations, 4 mutL homolog 1 mutations, 3 KRAS mutations, and 3 MET proto-oncogene mutations. These researchers also discovered two anaplastic lymphoma kinase (ALK) rearrangements, including glutamine-fructose-6-phosphate transaminase 1 (GFPT1)-ALK and echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusions. Interestingly, a metastatic MTC sample harboring the EML4-ALK fusion exhibited a remarkable response to an ALK inhibitor, crizotinib. These authors suggested that an EML4-ALK fusion in MTC may be a potential driver mutation and a suitable target for ALK inhibitors. They also concluded that the GFPT1-ALK fusion could be a potential target for molecular therapy [90]. Vaclavikova et al. performed RET genetic screening in 870 individuals—490 patients and 380 at-risk relatives. Fifty-eight families had RET germline mutations, however six families with hereditary forms of MTC had no mutation in all RET exons. They also analyzed six index patients of these families by means of the Trusight Cancer Sequencing Panel (Illumina, San Diego, CA, USA) which targeted 94 genes related to cancer. Analysis of these genes has identified very rare variants in the RHBDF2, XPA, MET, CHEK2, APC, and EZH2 genes. In addition, three patients were discovered with a pathogenic polymorphism in HNF1A [91]. Although several mutations were identified in MTCs by the NGS assay, no relevant therapeutic target has been discovered, and further investigation is needed to improve the treatment of MTC tumors.
5 Approved Tyrosine Kinase Inhibitors
5.1 Vandetanib
Vandetanib (4-anilinoquinazoline) is a multi-tyrosine kinase inhibitor (TKI) of RET, VEGFR-2, VEGFR-3, and EGFR. Phase I evaluation in patients with solid tumors showed that vandetanib monotherapy (up to 300 mg/day) was generally well tolerated, but no patients achieved an objective complete or partial response according to the Response Evaluation Criteria In Solid Tumors (RECIST) criteria. The percentage of patients with stable disease (SD) ranged from 12.5 to 50.0% [92]. At therapeutic doses, vandetanib can inhibit proliferation, survival, and angiogenesis in tumors, without toxicity to the tumor or endothelial cells [93]. The side effects of vandetanib include rash (especially photosensitivity), diarrhea, fatigue, nausea, and alteration in electrocardiogram [92]. According to the results of some investigations, MTC cells may escape from RET blockade through compensatory overstimulation of EGFR [94]; however, it has been reported that vandetanib, by double inhibition of RET and EGFR, can overcome the risk of this phenomenon [95]. In a phase II study, 30 adult patients with unresectable, progressive, or metastatic hereditary MTC received vandetanib 300 mg/day. Objective PRs were observed in 20% of patients and the median duration of PR was 10.2 months. Additionally, 53% of patients had SD for a median of 24 weeks [96]. In a randomized, phase III trial of vandetanib, patients were randomly assigned (2:1) to receive vandetanib (300 mg/day) or placebo. PRs were reported in 45% of patients, but no patients had SD [97]. The data reported in this phase III trial led to the approval of vandetanib by the US FDA in April 2011 [98]. Vandetanib is a first-line treatment for patients with unresectable symptomatic distant metastases, as well as a treatment of choice for advanced symptomatic metastatic disease, and is expected to be an important addition to the formulary of health plans that provide prescription drug benefits [99]. It has been reported that patients treated with vandetanib experienced a significant improvement in progression-free survival (PFS); however, no difference was observed in the overall survival (OS) of vandetanib-treated patients [100]. This drug is available as 100 and 300 mg tablets, with a recommended dose of 300 mg orally once daily. Vandetanib therapy should be continued until unacceptable toxicity occurs or the patient is no longer clinically benefiting from therapy [99]. Vandetanib is contraindicated in patients with congenital long-QT syndrome. In addition, vandetanib must be prohibited in patients with a history of torsades de pointes (TdP) and must not be initiated in patients with a baseline QT interval, Fridericia (QTcF interval) >450 ms [101]. In the US, vandetanib should only be prescribed by physicians after completion of a vandetanib Risk Evaluation and Mitigation Strategy (REMS) program because of a 178 black-box warning for QTc prolongation [94].
5.2 Cabozantinib
Cabozantinib is a small molecule that inhibits MET, VEGFR-2, RET, KIT, Flt-3, and Tie-2 [102]. By dual inhibition of VEGF and MET inhibition, cabozantinib can prevent MET pathway escape [103]. In a phase I trial of patients with MTC, cabozantinib induced a PR in 29% of patients, while 41% had SD for more than 6 months. To investigate two different schedules of administration and formulations of cabozantinib, patients were assigned to 13 dose levels; however, responses occurred most commonly at the 175 mg/day dose level. Adverse effects included aspartate aminotransferase/alanine aminotransferase (AST/ALT) elevation, fatigue, diarrhea, appetite loss, weight loss, stomatitis, hair hypopigmentation, hand–foot dermatitis, and hypertension [104]. Due to these encouraging findings, a multicenter, randomized, controlled, phase III trial (EXAM) was conducted where patients with MTC were randomized to cabozantinib (140 mg/day) or placebo in a 2:1 distribution (n = 330). In total, 312 patients (95%) could be assessed for tumor response. PR was 28% in the cabozantinib arm and 0% in the placebo arm, and the median estimated duration of response was 14.6 months. Ninety-five percent of cabozantinib-treated patients with measurable disease at baseline and at least one post-baseline assessment had a significant decrease in tumor lesion size when compared with placebo-treated patients [105]. These results led to the approval of the drug by the FDA in November 2012 for patients with progressive MTC [106]. The results of the EXAM study apparently indicated that cabozantinib constituted an effective treatment choice in patients with advanced, progressive MTC, both as first- and second-line treatment as this drug increased the time to tumor progression. Moreover, in a subgroup of patients with either germinal or sporadic tumor RET M918T mutation, the drug extended OS [107]. Comparison of the ratio of median PFS between cabozantinib and vandetanib showed that cabozantinib had greater benefit than vandetanib. Of note, one remarkable difference between the two drugs is that the cabozantinib-treated patients have no significant QTc interval prolongation, therefore cabozantinib should be the drug of choice to treat patients with a history of heart disease, arrhythmia, or QT prolongation [108]. The recommended starting dose of cabozantinib is 140 mg daily, with dose reductions to adjust for tolerability; efficacy data for a lower starting dose are unavailable. Although not mandated in its approval, safety monitoring during therapy should include periodic assessment of electrolytes, calcium, and thyroid-stimulating hormone [109].
6 Clinical Trials: TKIs
In the previous section we showed that there are different tyrosine kinases and pathways, which are abnormally activated or altered in MTC cells. Over the past decade various therapeutic agents have been studied for advanced MTC, the most promising results have arisen from the investigation of TKIs, which generally bind to ATP binding sites of these kinases. Nonetheless, inhibition of only one receptor or molecule in a pathway may not be effective and may cause the activation of other tyrosine kinases. Therefore, simultaneous inhibition of different activated tyrosine kinases may be the best way to approach MTC [94, 110]. In this section, we focus on the most common drugs that are in different phases of clinical trials (see Table 2 for a summary of these TKIs).
6.1 Sorafenib
Sorafenib was initially developed to target the rapidly accelerated fibrosarcoma (Raf) family of kinases, mainly B-Raf and C-Raf, but can also inhibit VEGFR-2, platelet-derived growth factor receptor (PDGFR)-β, c-Kit, FLT3, and RET. Sorafenib is particularly known as a potent inhibitor of RET kinase [111]. Sorafenib has mainly cytostatic activity, however the drug can also exert a proapoptotic effect. It has been reported that sorafenib could decrease tumor growth in nude mice with xenograft tumors derived from MTC cell lines [112]. In four phase I trials exploring different doses of sorafenib, the optimal therapeutic dose was found to be 400 mg twice daily. The most common adverse events included hand–foot syndrome, rash, fatigue, diarrhea, and hypertension. Antitumor effects of sorafenib was revealed by inducing disease stabilization in patients with refractory tumors [113]. Koh et al. conducted a study to identify potential pathways of escape from sorafenib at subtherapeutic concentrations. They suggested that the lack of change in total Erk expression during all treatments was due to escape from sorafenib signaling inhibition [114]. In an open-label, phase II study, sorafenib (400 mg twice daily) was administered to patients with advanced MTC. In this study, 16 or 25 patients were enrolled into arms A (hereditary) and B (sporadic); however, Arm A was prematurely terminated due to a slow accrual of patients. Of 16 patients treated in arm B, one achieved PR (6%), 14 had SD (80%), and one could not be evaluated [115]. Another phase II clinical trial was conducted to evaluate the tolerability and efficacy of sorafenib in 15 patients with advanced MTC and 19 patients with differentiated thyroid carcinoma (DTC) with non-radioiodine avid disease in a UK-based population. These patients were treated with sorafenib 400 mg twice daily until disease progression occurred. Response rate (RR) for MTC at 6 and 12 months was 13 and 25%, respectively. These data show that responses in MTC can occur beyond 6 months. For the radiologic RR, at 12 months PR was 21% and SD was 65%, but for the biochemical response, PR and SD were 48 and 11%, respectively. Interestingly, the PR was observed in RET mutation-negative MTC patients rather than RET mutation-positive MTC patients. This suggests that in vivo effects of sorafenib may not be RET inhibitory [116].
6.2 Tipifarnib
Tipifarnib inhibits farnesylation of RAS and other proteins. Farnesyltransferase catalyzes farnesylation of the RAS oncoprotein, leading to anchorage of RAS to the cell membrane. Substantial experimental evidence has linked farnesylation of Ras with malignancy. Farnesyltransferase inhibitors (FTIs) are a class of anticancer agents that can arrest malignant alterations with tolerable toxicity to normal cells. In addition, FTIs may inhibit other farnesylated proteins such as RhoB, a small GTPase, which has apoptotic and antineoplastic responses [117]. Two phase I clinical trials have been conducted to evaluate the combination of sorafenib and tipifarnib in advanced malignancies such as MTC. In one study, this combination was used in 15 patients with thyroid cancer, six of whom had MTC. Three (50%) of the six MTC patients who reached first restaging had a PR, lasting 12–26 months, whereas three (50%) had minor regressions (SD) lasting 12–16 months. In this study, a standard 3 + 3 dose-escalation design was used. Each cycle consisted of 28 days of sorafenib and 21 days of tipifarnib. The authors showed that the combination of sorafenib and tipifarnib is well tolerated at doses up to and including sorafenib 400 mg every morning/200 mg every afternoon orally once daily, and tipifarnib 100 mg orally twice daily. The most common side effects were grade 1–2 rash, hyperglycemia, and diarrhea [118]. The other phase I study was conducted to test this combination in 35 differentiated and MTC patients. PR in MTC patients was 50%, and 50% of these patients had SD. The maximum tolerated dose (MTD) was sorafenib 600 mg/tipifarnib 200 mg (split dosing). These drugs were administered for 21 days and repeated every 28 days. Major dose-limiting toxicities in this cohort were grade 1–3 rash, while other common skin toxicities were grade 1–2 hand–foot skin reaction [119].
6.3 Sunitinib
Sunitinib is an oral small-molecule TKI of all four VEGFRs, PDGFR, KIT, and RET [120]. Primary results from a phase II trial in advanced DTC or MTC patients showed PR in 13% of 31 DTC patients, and SD in 68% of DTC patients and 83% of MTC patients. Treatment consisted of 6-week cycles of sunitinib malate 50 mg daily on a 4-week on/2-week off schedule [121]. Results from another phase II study that enrolled patients with progressive MTC also reported responses. Of 23 evaluable patients, 35% reached PR and 57% had SD, with a median response duration of 37 and 32 weeks, respectively. Patients were treated with sunitinib 50 mg at a 4-/2-week schedule [122]. Recently, preliminary results from the other trial evaluating continuous administration of sunitinib (37.5 mg/day) were reported. Of 33 patients with 18F-fluorodeoxyglucose-positron emission tomography (FDG-PET)-avid metastatic thyroid cancer (23 with DTC, 7 with MTC), 29 were evaluable for response: 7% complete response (lasting at least 9 months), 25% PR, and 48% SD. Adverse effects included fatigue, diarrhea, palmar-plantar erythrodysesthesia, neutropenia, and hypertension [123].
6.4 Axitinib
Axitinib is an oral inhibitor that effectively blocks VEGFR1, 2, and 3 [124]. Sixty patients with advanced thyroid cancer [PTC, follicular (FTC), anaplastic (ATC), or MTC], which was not controlled by 131I, were enrolled into a phase II trial to receive axitinib orally (starting dose, 5 mg twice daily). Of these patients, 25% were not eligible for response assessment; however, the overall PR rate was 30% in an intent-to-treat analysis. Of 11 patients with MTC, 18% achieved PR and 27% had SD. Responses were observed in patients despite previous treatments with a number of chemotherapeutic agents. Median PFS was 18 months. Common side effects included fatigue, stomatitis, proteinuria, diarrhea, hypertension, and nausea [125].
6.5 Motesanib
Motesanib is a small molecule that can inhibit VEGFRs, PDGFR, and Kit [126]. In cellular phosphorylation assays, motesanib strongly inhibited wild-type RET, but had limited activity on mutant RET. In vivo, motesanib inhibited growth of xenografts of TT cells carrying the C634W RET mutation [127]. It has been reported that motesanib antagonizes MDR by inhibiting the efflux activity of the ATP-binding cassette subfamily B member 1 (ABCB1) transporter by binding to the transmembrane part of this protein [128]. In a phase I study, the safety, pharmacokinetics, and pharmacodynamics of motesanib in patients with refractory advanced solid tumors, including PTC, FTC and MTC, were assessed. The drug was used at an escalation schedule of 50–175 mg once daily or 25 mg twice daily for the first 21 days of a 28-day cycle. The 125 mg once-daily dose was also administered continuously. The MTD was 125 mg once-daily. Of the 67 assessable patients, 7% had a PR and 49% had SD, while, of the responders, three had thyroid cancer (MTC, PTC, and FTC). The most adverse events included fatigue, nausea, diarrhea, and hypertension [129]. These results led to a phase II study to investigate the efficacy and tolerability of motesanib (125 mg/day orally for up to 48 weeks) in a cohort of patients with advanced or symptomatic MTC. Of 91 enrolled patients who were treated with motesanib, 2% achieved objective response and 81% had SD. The most common drug-related side effects were diarrhea, fatigue, hypothyroidism, hypertension, and anorexia [130].
6.6 Imatinib
Imatinib is a small molecule that inhibits c-KIT, RET autophosphorylation, and RET-mediated cell growth [131]. A phase II study was conducted to examine the effect of imatinib (600 mg/day, with a possible dose increase to 800 mg in case of progression) in 15 patients with disseminated MTC. No objective responses were observed. Four patients (26.6%) had SD over 24 months, three patients stopped treatment due to toxicity, and in four cases the dose of imatinib was reduced because of toxic effects (fatigue, nausea, rash, malaise, laryngeal swelling) [132]. In an open-label clinical trial, imatinib mesylate (600 mg/day) was administered in nine patients (eight with sporadic MTC and one with hereditary MTC) with unresectable, measurable, progressive metastases. SD lasting up to 6 months was observed in five patients (55.5%) and for up to 12 months in one patient (11%); no PR occurred [133].
6.7 Gefitinib
Gefitinib is a potent inhibitor of EGFR. The drug was first used for the therapy of non small cell lung carcinoma [134]. In a phase II trial, 27 patients with radioiodine-refractory or metastatic thyroid cancer received gifitinib (250 mg daily). The cohort included patients with PTC (41%), FTC (22%), ATC (19%), MTC (15%), and Hürthle cell carcinoma (4%). No partial or complete responses were observed in the 25 evaluable patients. The SD rate at 3, 6, and 12 months was 48, 24, and 12%, respectively, and the most common toxic events were rash, diarrhea, nausea, and anorexia [135].
6.8 Lenvatinib
Lenvatinib is a small molecule that can inhibit both VEGFR-2 and VEGFR-3 [136]. A phase II clinical trial was conducted to assess the effect of lenvatinib on tumor growth in MTC patients, in which 59 patients with unresectable advanced MTC were treated with lenvatinib (24 mg daily, 28-day cycles) until disease progression, unmanageable toxicity, withdrawal, or death. PR was 36, and 44% of patients had SD. The most common adverse events were diarrhea, hypertension, decreased appetite, fatigue, dysphagia, and increased ALT levels [137]. Another open-label, multicenter, phase II study was initiated in Japan in 60 patients, including 22 DTC, 4 MTC, and 9 ATC patients. Patients received lenvatinib at a starting dose of 24 mg/day in 28-day cycles. PR based on investigator evaluations was 47.6% in RR-DTC, 25.0% in MTC, and 33.3% in ATC, respectively. Side effects included hypertension, palmar-plantar erythrodysesthesia syndrome, fatigue, decreased appetite, proteinuria, stomatitis, diarrhea, nausea and dysphonia [138]. In their study of nine patients (three females, 61 ± 8 years) with progressive iodine-refractory DTC starting on lenvatinib, Werner et al. reported that after a mean follow-up of 19 months, only three of nine patients have stopped treatment [139].
6.9 Pazopanib
Pazopanib is a small molecule TKI that targets VEGFR-1, -2, and -3, PDGFR-α and -β, and c-Kit. It has no inhibitory effects on oncogenic kinases RET, RET/PTC, or BRAF; therefore, it is believed that its primary action in thyroid cancers is antiangiogenic. Thirty-five patients with advanced and metastatic MTC were enrolled in a multicenter, phase II trial to assess tumor response to pazopanib (800 mg/day). Five patients (14.3%) reached PR evaluation criteria in solid tumor responses, and 20 patients (57%) had SD. Side effects included treatment-requiring hypertension, fatigue, diarrhea, and abnormal liver tests. Three patients (8.6%) discontinued therapy due to adverse events [140].
7 Conclusions
Although surgery is now the main treatment option for advanced MTCs, understanding the molecular mechanisms of these tumors is the best way to utilize non-invasive methods for treating patients. Nonetheless, molecular-targeted therapies are not totally harmless and may reduce patients’ quality of life. For example, two drugs (vandetanib and cabozantinib), which are approved for the treatment of advanced MTC, also have adverse effects on patients’ quality of life. Intrinsic or secondary drug resistance is a common and significant challenge if monotherapy is used. Accordingly, the drug combination approach utilizing different agents with different targets may appear an effective strategy to avoid drug resistance.
References
Ball DW. Medullary thyroid carcinoma. Thyroid cancer. Berlin: Springer; 2000. p. 365–81.
Niccoli-Sire P, Conte-Devolx B (eds). Medullary thyroid carcinoma. Annales d’endocrinologie; 2007.
Jaquet J. Ein fall von metastasierenden amyloidtumoren (lymphosarkom). Virchows Arch. 1906;185(2):251–68.
Hazard JB, Hawk WA, Crile JRG. Medullary (solid) carcinoma of the thyroid—a clinicopathologic entity. J Clin Endocrinol Metab. 1959;19(1):152–61.
Schmid KW. Histopathology of C cells and medullary thyroid carcinoma. Medullary thyroid carcinoma. Berlin: Springer; 2015. p. 41–60.
Williams E. Histogenesis of medullary carcinoma of the thyroid. J Clin Pathol. 1966;19(2):114–8.
Bussolati G, Pearse A. Immunofluorescent localization of calcitonin in the ‘C’ cells of pig and dog thyroid. J Endocrinol. 1967;37(2):205.
Tashjian AH Jr, Melvin KE. Medullary carcinoma of the thyroid gland: studies of thyrocalcitonin in plasma and tumor extracts. N Engl J Med. 1968;279(6):279–83.
Melvin KE, Miller HH, Tashjian AH Jr. Early diagnosis of medullary carcinoma of the thyroid gland by means of calcitonin assay. N Engl J Med. 1971;285(20):1115–20.
Hu MI, Ying AK, Jimenez C. Update on medullary thyroid cancer. Endocrinol Metab Clin North Am. 2014;43(2):423–42.
Ernani V, Kumar M, Chen AY, Owonikoko TK. Systemic treatment and management approaches for medullary thyroid cancer. Cancer Treatm Rev. 2016;50:89–98.
Pacini F, Castagna M, Cipri C, Schlumberger M. Medullary thyroid carcinoma. Clin Oncol. 2010;22(6):475–85.
Sippel RS, Kunnimalaiyaan M, Chen H. Current management of medullary thyroid cancer. Oncologist. 2008;13(5):539–47.
Marquard J, Eng C. Multiple endocrine neoplasia type 2. GeneReviews. 2015.
Moley JF. Medullary thyroid carcinoma. Curr Treatm Options Oncol. 2003;4(4):339–47.
Sheikholeslami S, Yeganeh MZ, Rad LH, Ghadaksaz HG, Hedayati M. Haplotype Frequency of G691S/S904S in the RET proto-onco-gene in patients with medullary thyroid carcinoma. Iran J Publ Health. 2014;43(2):235.
Kebebew E, Ituarte PH, Siperstein AE, Duh QY, Clark OH. Medullary thyroid carcinoma. Cancer. 2000;88(5):1139–48.
Barbosa SLS, Rodien P, Leboulleux S, Niccoli-Sire P, Kraimps JL, Caron P, et al. Ectopic adrenocorticotropic hormone-syndrome in medullary carcinoma of the thyroid: a retrospective analysis and review of the literature. Thyroid. 2005;15(6):618–23.
Wohllk N, Schweizer H, Erlic Z, Schmid KW, Walz MK, Raue F, et al. Multiple endocrine neoplasia type 2. Best Pract Res Clin Endocrinol Metab. 2010;24(3):371–87.
Romei C, Mariotti S, Fugazzola L, Taccaliti A, Pacini F, Opocher G, et al. Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol. 2010;163(2):301–8.
DeLellis R, Wolfe H, Gagel R, Feldman Z, Miller H, Gang D, et al. Adrenal medullary hyperplasia. A morphometric analysis in patients with familial medullary thyroid carcinoma. Am J Pathol. 1976;83(1):177.
Carney J, Sizemore G, Tyce G. Bilateral adrenal medullary hyperplasia in multiple endocrine neoplasia, type 2: the precursor of bilateral pheochromocytoma. Mayo Cli Proc. 1975;50(1):3–10.
Gagel RF, Tashjian AH Jr, Cummings T, Papathanasopoulos N, Kaplan MM, DeLellis RA, et al. The clinical outcome of prospective screening for multiple endocrine neoplasia type 2a. N Engl J Med. 1988;318(8):478–84.
Verdy M, Weber AM, Roy CC, Morin CL, Cadotte M, Brochu P. Hirschsprung’s disease in a family with multiple endocrine neoplasia type 2. J Pediatr Gastroenterol Nutr. 1982;1(4):603–8.
Carney J, Sizemore G, Hayles A. Multiple endocrine neoplasia, type 2b. Pathobiol Annu. 1977;8:105–53.
Leboulleux S, Travagli J, Caillou B, Laplanche A, Bidart J, Schlumberger M, et al. Medullary thyroid carcinoma as part of a multiple endocrine neoplasia type 2B syndrome. Cancer. 2002;94(1):44–50.
Nosé V. Familial thyroid cancer: a review. Modern Pathol. 2011;24:S19–33.
Elisei R, Romei C, Cosci B, Agate L, Bottici V, Molinaro E, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab. 2007;92(12):4725–9.
Pasini B, Hofstra R, Yin L, Bocciardi R, Santamaria G, Grootscholten PM, et al. The physical map of the human RET proto-oncogene. Oncogene. 1995;11(9):1737–43.
Anders J, Kjær S, Ibáñez CF. Molecular modeling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin-like domains and a calcium-binding site. J Biol Chem. 2001;276(38):35808–17.
Tsui-Pierchala BA, Milbrandt J, Johnson EM. NGF utilizes c-Ret via a novel GFL-independent, inter-RTK signaling mechanism to maintain the trophic status of mature sympathetic neurons. Neuron. 2002;33(2):261–73.
Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nature Rev Neurosci. 2002;3(5):383–94.
Ichihara M, Murakumo Y, Takahashi M. RET and neuroendocrine tumors. Cancer Lett. 2004;204(2):197–211.
Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985;42(2):581–8.
Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Human Mol Genet. 1993;2(7):851–6.
Mulligan LM, Kwok J, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363(6428):458–60.
Carlson KM, Dou S, Chi D, Scavarda N, Toshima K, Jackson CE, et al. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci. 1994;91(4):1579–83.
Eng C, SmIth DP, MullIgan LM, Nagal MA, Healey CS, Ponder MA, et al. Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Human Mol Genet. 1994;3(2):237–41.
Marsh DJ, Learoyd DL, Andrew SD, Krishnan L, Pojer R, Richardson AL, et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinoma. Clin Endocrinol. 1996;44(3):249–57.
Yeganeh MZ, Sheikholeslami S, Behbahani GD, Farashi S, Hedayati M. Skewed mutational spectrum of RET proto-oncogene Exon10 in Iranian patients with medullary thyroid carcinoma. Tumor Biol. 2015;36(7):5225–31.
Schilling T, Bürck J, Sinn HP, Clemens A, Otto HF, Höppner W, et al. Prognostic value of codon 918 (ATG → ACG) RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. Int J Cancer. 2001;95(1):62–6.
Elisei R, Cosci B, Romei C, Bottici V, Renzini G, Molinaro E, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 2008;93(3):682–7.
Moura M, Cavaco B, Pinto A, Domingues R, Santos J, Cid M, et al. Correlation of RET somatic mutations with clinicopathological features in sporadic medullary thyroid carcinomas. Br J Cancer. 2009;100(11):1777–83.
Santoro M, Melillo RM, Fusco A. RET/PTC activation in papillary thyroid carcinoma. Eur J Endocrinol Prize Lect Eur J Endocrinol. 2006;155(5):645–53.
Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nature Med. 2012;18(3):375–7.
Ballerini P, Struski S, Cresson C, Prade N, Toujani S, Deswarte C, et al. RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia. 2012;26(11):2384–9.
Coyle D, Friedmacher F, Puri P. The association between Hirschsprung’s disease and multiple endocrine neoplasia type 2a: a systematic review. Pediatr Surg Int. 2014;30(8):751–6.
Margraf RL, Crockett DK, Krautscheid PM, Seamons R, Calderon FR, Wittwer CT, et al. Multiple endocrine neoplasia type 2 RET protooncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Human Mutat. 2009;30(4):548–56.
Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, et al. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun. 1992;187(3):1579–86.
Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312(5):549–60.
Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358(19):2039–49.
Rodríguez-Antona C, Pallares J, Montero-Conde C, Inglada-Pérez L, Castelblanco E, Landa I, et al. Overexpression and activation of EGFR and VEGFR2 in medullary thyroid carcinomas is related to metastasis. Endocr Relat Cancer. 2010;17(1):7–16.
Bunone G, Vigneri P, Mariani L, Butó S, Collini P, Pilotti S, et al. Expression of angiogenesis stimulators and inhibitors in human thyroid tumors and correlation with clinical pathological features. Am J Pathol. 1999;155(6):1967–76.
Capp C, Wajner SM, Siqueira DR, Brasil BA, Meurer L, Maia AL. Increased expression of vascular endothelial growth factor and its receptors, VEGFR-1 and VEGFR-2, in medullary thyroid carcinoma. Thyroid. 2010;20(8):863–71.
Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Investig. 2007;117(8):2051.
Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res. 2003;284(1):31–53.
Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Exp Cell Res. 2003;284(1):99–110.
Vlahovic G, Crawford J. Activation of tyrosine kinases in cancer. Oncologist. 2003;8(6):531–8.
Croyle M, Akeno N, Knauf JA, Fabbro D, Chen X, Baumgartner JE, et al. RET/PTC-induced cell growth is mediated in part by epidermal growth factor receptor (EGFR) activation: evidence for molecular and functional interactions between RET and EGFR. Cancer Res. 2008;68(11):4183–91.
Bottaro DP, Rubin JS, Faletto DL, Chan A, Kmiecik TE, Woude GV, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251(4995):802–4.
Corso S, Migliore C, Ghiso E, De Rosa G, Comoglio P, Giordano S. Silencing the MET oncogene leads to regression of experimental tumors and metastases. Oncogene. 2008;27(5):684–93.
Di Renzo M, Narsimhan R, Olivero M, Bretti S, Giordano S, Medico E, et al. Expression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene. 1991;6(11):1997–2003.
Di Renzo MF, Olivero M, Giacomini A, Porte H, Chastre E, Mirossay L, et al. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res. 1995;1(2):147–54.
Di Renzo M, Olivero M, Serini G, Orlandi F, Pilotti S, Belfiore A, et al. Overexpression of the c-MET/HGF receptor in human thyroid carcinomas derived from the follicular epithelium. J Endocrinol Investig. 1995;18(2):134–9.
Di Renzo MF, Olivero M, Katsaros D, Crepaldi T, Gaglia P, Zola P, et al. Overexpression of the Met/HGF receptor in ovarian cancer. Int J Cancer. 1994;58(5):658–62.
Ramirez R, Hsu D, Patel A, Fenton C, Dinauer C, Tuttle RM, et al. Over-expression of hepatocyte growth factor/scatter factor (HGF/SF) and the HGF/SF receptor (cMET) are associated with a high risk of metastasis and recurrence for children and young adults with papillary thyroid carcinoma. Clin Endocrinol. 2000;53(5):635–44.
Prat M, Narsimhan RP, Crepaldi T, Rita Nicotra M, Natali PG, Comoglio PM. The receptor encoded by the human C-MET oncogene is expressed in hepatocytes, epithelial cells and solid tumors. Int J Cancer. 1991;49(3):323–8.
Liu C, Park M, Tsao M. Overexpression of c-met proto-oncogene but not epidermal growth factor receptor or c-erbB-2 in primary human colorectal carcinomas. Oncogene. 1992;7(1):181–5.
Jeffers M, Schmidt L, Nakaigawa N, Webb CP, Weirich G, Kishida T, et al. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci. 1997;94(21):11445–50.
Schmidt L, Duh F-M, Chen F, Kishida T, Glenn G, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nature Genet. 1997;16(1):68–73.
Papotti M, Olivero M, Volante M, Negro F, Prat M, Comoglio PM, et al. Expression of hepatocyte growth factor (HGF) and its receptor (MET) in medullary carcinoma of the thyroid. Endocr Pathol. 2000;11(1):19–30.
Wasenius V-M, Hemmer S, Karjalainen-Lindsberg M-L, Nupponen NN, Franssila K, Joensuu H. MET receptor tyrosine kinase sequence alterations in differentiated thyroid carcinoma. Am J Surg Pathol. 2005;29(4):544–9.
Cassinelli G, Favini E, Degl’Innocenti D, Salvi A, De Petro G, Pierotti MA, et al. RET/PTC1-driven neoplastic transformation and proinvasive phenotype of human thyrocytes involve Met induction and β-catenin nuclear translocation. Neoplasia. 2009;11(1):10–21.
Ivan M, Bond JA, Prat M, Comoglio PM, Wynford-Thomas D. Activated ras and ret oncogenes induce over-expression of c-met (hepatocyte growth factor receptor) in human thyroid epithelial cells. Oncogene. 1997;14(20):2417–23.
Eswarakumar V, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16(2):139–49.
St. Bernard R, Zheng L, Liu W, Winer D, Asa SL, Ezzat S. Fibroblast growth factor receptors as molecular targets in thyroid carcinoma. Endocrinology. 2005;146(3):1145–53.
Ezzat S, Huang P, Dackiw A, Asa SL. Dual inhibition of RET and FGFR4 restrains medullary thyroid cancer cell growth. Clin Cancer Res. 2005;11(3):1336–41.
Rajalingam K, Schreck R, Rapp UR, Albert Š. Ras oncogenes and their downstream targets. Biochimica et biophysica acta (BBA). Mol Cell Res. 2007;1773(8):1177–95.
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nature Rev Cancer. 2011;11(11):761–74.
Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457–67.
Moura MM, Cavaco BM, Leite V. RAS proto-oncogene in medullary thyroid carcinoma. Endocr Relat Cancer. 2015;22(5):R235–52.
Ciampi R, Mian C, Fugazzola L, Cosci B, Romei C, Barollo S, et al. Evidence of a low prevalence of RAS mutations in a large medullary thyroid cancer series. Thyroid. 2013;23(1):50–7.
Agrawal N, Jiao Y, Sausen M, Leary R, Bettegowda C, Roberts NJ, et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab. 2012;98(2):E364–9.
D’Argenio V. Molecular alterations in human genetic diseases through next generation sequencing technologies [thesis]. European School of Molecular Medicine (SEMM) Universita’ Degli Studi Di Napoli Federico II; 2015.
Nikiforova MN, Wald AI, Roy S, Durso MB, Nikiforov YE. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J Clin Endocrinol Metab. 2013;98(11):E1852–60.
Moura MM, Cavaco BM, Pinto AE, Leite V. High prevalence of RAS mutations in RET-negative sporadic medullary thyroid carcinomas. J Clin Endocrinol Metab. 2011;96(5):E863–8.
Wei S, LiVolsi VA, Montone KT, Morrissette JJ, Baloch ZW. Detection of molecular alterations in medullary thyroid carcinoma using next-generation sequencing: an institutional experience. Endocr Pathol. 2016;27(4):359–62.
Simbolo M, Mian C, Barollo S, Fassan M, Mafficini A, Neves D, et al. High-throughput mutation profiling improves diagnostic stratification of sporadic medullary thyroid carcinomas. Virchows Arch. 2014;465(1):73–8.
Grubbs EG, Ng PK-S, Bui J, Busaidy NL, Chen K, Lee JE, et al. RET fusion as a novel driver of medullary thyroid carcinoma. J Clin Endocrinol Metab. 2015;100(3):788–93.
Ji JH, Oh YL, Hong M, Yun JW, Lee H-W, Kim D, et al. Identification of driving ALK fusion genes and genomic landscape of medullary thyroid cancer. PLoS Genet. 2015;11(8):e1005467.
Vaclavikova E, Dvorakova S, Sykorova V, Vcelak J, Halkova T, Vlcek P, et al. Search for new candidate genes in RET mutation-negative families with hereditary medullary thyroid carcinoma using next generation sequencing. Endocr Abstr. 2015;37(OC6):5.
Holden S, Eckhardt S, Basser R, De Boer R, Rischin D, Green M, et al. Clinical evaluation of ZD6474, an orally active inhibitor of VEGF and EGF receptor signaling, in patients with solid, malignant tumors. Ann Oncol. 2005;16(8):1391–7.
Herbst RS, Heymach JV, O’Reilly MS, Onn A, Ryan AJ. Vandetanib (ZD6474): an orally available receptor tyrosine kinase inhibitor that selectively targets pathways critical for tumor growth and angiogenesis. Exp Opin Investig Drugs. 2007;16(2):239–49.
Vitagliano D, De Falco V, Tamburrino A, Coluzzi S, Troncone G, Chiappetta G, et al. The tyrosine kinase inhibitor ZD6474 blocks proliferation of RET mutant medullary thyroid carcinoma cells. Endocr Relat Cancer. 2011;18(1):1–11.
Gomez K, Varghese J, Jiménez C. Medullary thyroid carcinoma: molecular signaling pathways and emerging therapies. J Thyroid Res. 2011;2011:815826. doi:10.4061/2011/815826.
Wells SA, Gosnell JE, Gagel RF, Moley J, Pfister D, Sosa JA, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28(5):767–72.
Wells SA, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30(2):134–41.
Degrauwe N, Sosa JA, Roman S, Deshpande HA. Vandetanib for the treatment of metastatic medullary thyroid cancer. Clin Med Insights Oncol. 2012;6:243.
Cooper MR, Yi SY, Alghamdi W, Shaheen DJ, Steinberg M. Vandetanib for the treatment of medullary thyroid carcinoma. Ann Pharmacother. 2014;48(3):387–94.
Fallahi P, Ferrari SM, Baldini E, Biricotti M, Ulisse S, Materazzi G, et al. The safety and efficacy of vandetanib in the treatment of progressive medullary thyroid cancer. Exp Rev Anticancer Ther. 2016;16(11):1109–18.
Tsang VH, Robinson BG, Learoyd DL. The safety of vandetanib for the treatment of thyroid cancer. Exp Opin Drug Saf. 2016;15(8):1107–13.
Eder JP, Woude GFV, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15(7):2207–14.
Hart CD, De Boer RH. Profile of cabozantinib and its potential in the treatment of advanced medullary thyroid cancer. Onco Targets Ther. 2013;6:1–7.
Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29(19):2660–6.
Elisei R, Schlumberger MJ, Müller SP, Schöffski P, Brose MS, Shah MH, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31(29):3639–46.
Nix NM, Braun K. Cabozantinib for the treatment of metastatic medullary thyroid carcinoma. J Adv Pract Oncol. 2014;5(1):47.
Krajewska J, Olczyk T, Jarzab B. Cabozantinib for the treatment of progressive metastatic medullary thyroid cancer. Exp Rev Clin Pharmacol. 2016;9(1):69–79.
Colombo JR, Wein RO. Cabozantinib for progressive metastatic medullary thyroid cancer: a review. Ther Clin Risk Manag. 2014;10:395.
Sherman IS, Douglas SR, Jean EM. Medullary thyroid cancer: chemotherapy and immunotherapy. 2017. Available at: http://www.uptodate.com.
Ocana A, Amir E, Seruga B, Pandiella A. Do we have to change the way targeted drugs are developed? J Clin Oncol. 2010;28(24):e420–1.
Hong D, Ye L, Gagel R, Chintala L, El Naggar AK, Wright J, et al. Medullary thyroid cancer: targeting the RET kinase pathway with sorafenib/tipifarnib. Mol Cancer Ther. 2008;7(5):1001–6.
Carlomagno F, Anaganti S, Guida T, Salvatore G, Troncone G, Wilhelm SM, et al. BAY 43-9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst. 2006;98(5):326–34.
Strumberg D, Clark JW, Awada A, Moore MJ, Richly H, Hendlisz A, et al. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist. 2007;12(4):426–37.
Koh YW, Shah MH, Agarwal K, McCarty SK, Koo BS, Brendel VJ, et al. Sorafenib and Mek inhibition is synergistic in medullary thyroid carcinoma in vitro. Endocr Relat Cancer. 2012;19(1):29–38.
Lam ET, Ringel MD, Kloos RT, Prior TW, Knopp MV, Liang J, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010;28(14):2323–30.
Ahmed M, Barbachano Y, Riddell A, Hickey J, Newbold KL, Viros A, et al. Analysis of the efficacy and toxicity of sorafenib in thyroid cancer: a phase II study in a UK based population. Eur J Endocrinol. 2011;165(2):315–22.
Izbicka E, Campos D, Carrizales G, Patnaik A. Biomarkers of anticancer activity of R115777 (Tipifarnib, Zarnestra™) in human breast cancer models in vitro. Anticancer Res. 2005;25(5):3215–23.
Hong DS, Sebti SM, Newman RA, Blaskovich MA, Ye L, Gagel RF, et al. Phase I trial of a combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in advanced malignancies. Clin Cancer Res. 2009;15(22):7061–8.
Cabanillas M, Kurzrock R, Sherman S, Tsimberidou A, Waguespack S, Naing A, Busaidy N, Gagel R, Wright JJ, Hong DS. Phase I trial of combination sorafenib and tipifarnib: The experience in advanced differentiated thyroid cancer (DTC) and medullary thyroid cancer (MTC). J Clin Oncol. 2010;28(15_suppl):5586.
Mena AC, Pulido EG, Guillen-Ponce C. Understanding the molecular-based mechanism of action of the tyrosine kinase inhibitor: sunitinib. Anticancer Drugs. 2010;21:S3–11.
Cohen EE, Needles BM, Cullen KJ, Wong SJ, Wade III JL, Ivy SP, Villaflor VM, Seiwert TY, Nichols K, Vokes EE. Phase 2 study of sunitinib in refractory thyroid cancer. J Clin Oncol. 2008;26(15_suppl):6025.
De Souza JA, Busaidy N, Zimrin A, Seiwert TY, Villaflor VM, Poluru KB, Reddy PL, Nam J, Vokes EE, Cohen EE. Phase II trial of sunitinib in medullary thyroid cancer (MTC). J Clin Oncol. 2010;28(15_suppl):5504.
Carr L, Goulart B, Martins R, Keith E, Kell E, Wallace S, et al. (eds). Phase II trial of continuous dosing of sunitinib in advanced, FDG-PET avid, medullary thyroid carcinoma (MTC) and well-differentiated thyroid cancer (WDTC). ASCO Annu Meet Proc.; 2009.
Escudier B, Gore M. Axitinib for the management of metastatic renal cell carcinoma. Drugs R & D. 2011;11(2):113–26.
Cohen EE, Rosen LS, Vokes EE, Kies MS, Forastiere AA, Worden FP, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol. 2008;26(29):4708–13.
Coxon A, Ziegler B, Kaufman S, Xu M, Wang H, Weishuhn D, et al. Antitumor activity of motesanib alone and in combination with cisplatin or docetaxel in multiple human non-small-cell lung cancer xenograft models. Mol Cancer. 2012;11(1):1.
Coxon A, Bready J, Kaufman S, Estrada J, Osgood T, Canon J, et al. Anti-tumor activity of motesanib in a medullary thyroid cancer model. J Endocrinol Investig. 2012;35(2):181–90.
Wang Y-J, Kathawala RJ, Zhang Y-K, Patel A, Kumar P, Shukla S, et al. Motesanib (AMG706), a potent multikinase inhibitor, antagonizes multidrug resistance by inhibiting the efflux activity of the ABCB1. Biochem Pharmacol. 2014;90(4):367–78.
Rosen LS, Kurzrock R, Mulay M, Van Vugt A, Purdom M, Ng C, et al. Safety, pharmacokinetics, and efficacy of AMG 706, an oral multikinase inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2007;25(17):2369–76.
Schlumberger MJ, Elisei R, Bastholt L, Wirth LJ, Martins RG, Locati LD, et al. Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. J Clin Oncol. 2009;27(23):3794–801.
de Groot J, Menacho IP, Schepers H, Drenth-Diephuis L, Osinga J, Plukker JTM, et al. Cellular effects of imatinib on medullary thyroid cancer cells harboring multiple endocrine neoplasia Type 2A and 2B associated RET mutations. Surgery. 2006;139(6):806–14.
De Groot J, Zonnenberg B, van Ufford-Mannesse PQ, De Vries M, Links T, Lips C, et al. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2007;92(9):3466–9.
Frank-Raue K, Fabel M, Delorme S, Haberkorn U, Raue F. Efficacy of imatinib mesylate in advanced medullary thyroid carcinoma. Eur J Endocrinol. 2007;157(2):215–20.
Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305(5687):1163–7.
Pennell NA, Daniels GH, Haddad RI, Ross DS, Evans T, Wirth LJ, et al. A phase II study of gefitinib in patients with advanced thyroid cancer. Thyroid. 2008;18(3):317–23.
Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14(17):5459–65.
Schlumberger M, Jarzab B, Cabanillas ME, Robinson B, Pacini F, Ball DW, et al. A phase II trial of the multitargeted tyrosine kinase inhibitor lenvatinib (E7080) in advanced medullary thyroid cancer. Clin Cancer Res. 2016;22(1):44–53.
Takahashi S, Tahara M, Kiyota N, Yamazaki T, Chayahara N, Nakano K, et al. 995PD pHASE II study of lenvatinib (LEN), a multi-targeted tyrosine kinase inhibitor, in patients (pts) with all histologic subtypes of advanced thyroid cancer (differentiated, medullary and anaplastic). Ann Oncol. 2014;25(Suppl 4):iv343–4.
Werner RA, Lückerath K, Schmid JS, Higuchi T, Kreissl MC, Grelle I, Reiners C, Buck AK, Lapa C. Thyroglobulin fluctuations in patients with iodine-refractory differentiated thyroid carcinoma on lenvatinib treatment–initial experience. Sci Rep. 2016;6:28081. doi:10.1038/srep28081.
Bible KC, Suman VJ, Molina JR, Smallridge RC, Maples WJ, Menefee ME, et al. A multicenter phase 2 trial of pazopanib in metastatic and progressive medullary thyroid carcinoma: MC057H. J Clin Endocrinol Metab. 2014;99(5):1687–93.
Acknowledgements
The authors are grateful to Dr. Javad Sharifi-Rad, Phytochemistry Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran, for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Sadegh Rajabi and Mehdi Hedayati have no conflict of interest.
Funding
The authors have no funding to declare.
Rights and permissions
About this article

Cite this article
Rajabi, S., Hedayati, M. Medullary Thyroid Cancer: Clinical Characteristics and New Insights into Therapeutic Strategies Targeting Tyrosine Kinases. Mol Diagn Ther 21, 607–620 (2017). https://doi.org/10.1007/s40291-017-0289-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-017-0289-5