
Abstract
The metabolic syndrome associates metabolic abnormalities such as insulin resistance and dyslipidemia with increased waist circumference and hypertension. It is a major public health concern, as its prevalence could soon reach 30% to 50% in developed countries. Aldosterone, a mineralocorticoid hormone classically involved in sodium balance regulation, is increased in patients with metabolic syndrome. Besides its classic actions, aldosterone and mineralocorticoid receptor (MR) activation affect glucose metabolism, inducing insulin resistance through various mechanisms that involve oxidative stress, inflammation, and downregulation of proteins involved in insulin signaling pathways. Aldosterone and MR signaling exert deleterious effects on the cardiovascular system and the kidney that influence the cardiovascular risk associated with metabolic syndrome. Salt load plays a major role in cardiovascular injury induced by aldosterone and MR signaling. Large multicenter, randomized clinical trials testing the beneficial effects of MR antagonists on cardiovascular events and mortality in patients with metabolic syndrome are needed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The metabolic syndrome includes a constellation of metabolic disturbances that reflect the increasing prevalence of obesity. Various diagnostic criteria have been proposed in the past 10 years by different organizations such as the World Health Organization in 1999, the European Group for the Study of Insulin Resistance in 1999, and the National Cholesterol Education Program Adult Treatment Panel III (ATP III) in 2001 [1]. A harmonization effort has resulted in the current definition of metabolic syndrome, which now includes an increased waist circumference, elevated triglycerides (≥1.7 mmol/L), reduced high-density lipoprotein (HDL) cholesterol level (≤1 mmol/L in males and ≤1.3 mmol/L in females), elevated blood pressure (systolic blood pressure ≥130 mm Hg or diastolic blood pressure ≥85 mm Hg), and elevated fasting glucose (≥100 mg/dL or 5.6 mmol/L) [2••].
The metabolic syndrome is cause for important public health concern. Its prevalence has increased over time. In the National Health and Examination Survey (NHANES) cohort, the prevalence between 1988 and 1994 was 29.2% using ATP III criteria, and it reached 34.6% between 1999 and 2002. The metabolic syndrome is associated with a well-recognized increase in cardiovascular risk [3]. A recent meta-analysis that included 87 studies demonstrated that patients with metabolic syndrome as defined by the 2001 and the 2004 revised National Cholesterol Education Program had a relative risk of cardiovascular disease of 2.35 (95% CI, 2.02–2.73) [4•].
Elevated plasma aldosterone values have been found in obese patients and in patients with the metabolic syndrome. Aldosterone is a mineralocorticoid hormone classically involved in sodium balance regulation, and it could contribute to the hypertension associated with the metabolic syndrome. Besides this action, aldosterone exerts deleterious effect on the kidney and the cardiovascular system that could contribute to the increase in cardiovascular risk associated with the metabolic syndrome [5]. A growing body of evidence has shown that aldosterone may also participate in the pathophysiology of other components of the metabolic syndrome, such as insulin resistance. The longitudinal follow-up of 2,292 participants of the Framingham Offspring Study underlined the importance of aldosterone in incident metabolic syndrome. Among the various biomarkers studied, aldosterone and plasminogen activator inhibitor-1 (PAI-1) were the only biomarkers independently associated with the occurrence of metabolic syndrome [6]. This review discusses the implications of aldosterone in the pathophysiology of the metabolic syndrome and in the increased cardiovascular risk observed in these patients.
Mineralocorticoid Receptor Expression and Aldosterone Effects
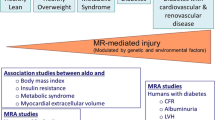
Aldosterone is a mineralocorticoid hormone mainly synthesized by the zona glomerulosa of the adrenal gland in response to angiotensin II, adrenocorticotropic hormone (ACTH), and hyperkalemia. Adrenal secretion of aldosterone stimulated by any of these agents is inhibited by natriuretic peptides such as the atrial natriuretic peptide [7]. Aldosterone is classically involved in transepithelial electrolyte transport of sodium and potassium in the distal convoluted tubule of the kidney via its action on the intracellular mineralocorticoid receptor (MR). More precisely, aldosterone, via MR, activates the apical epithelial sodium channel, ENaC, and the basolateral Na+K+ATPase [5]. MR expression is not restricted to the kidney but is widely distributed across a range of extraepithelial tissues such as heart, endothelial cells, vascular smooth muscle cells, and kidney mesangial cells and podocytes [5]. MR is also present in human adipocytes, preadipocytes and differentiated adipocytes, and the localization is nuclear and perinuclear [8•]. Besides its classic action at the level of the distal tubule of the kidney, aldosterone and MR signaling exert well-demonstrated deleterious effects on the heart, blood vessels, and kidneys, such as glomerulosclerosis and tubulointerstitial inflammation and fibrosis in the kidneys, and cardiovascular inflammation, fibrosis, and hypertrophic remodeling (extensively described elsewhere [5]) (Fig. 1).

Effects of aldosterone on metabolic syndrome. Increased plasma aldosterone levels are associated with obesity and metabolic syndrome. In turn, aldosterone has deleterious effects on the liver; effects on adipocytes and skeletal muscle that lead to insulin resistance; effects on the cardiovascular system that lead to inflammation, oxidative stress, hypertrophic remodeling, and endothelial dysfunction; and effects on the kidney that lead to glomerulosclerosis, tubulointerstitial inflammation and fibrosis, and podocyte dysfunction. The effects of aldosterone on lipid metabolism are more controversial. HDL—high-density lipoprotein
Interestingly, MR has the same level of affinity for aldosterone as for the glucocorticoids (GC) cortisol and corticosterone. Circulating GC levels are 1000-fold to 2000-fold higher than those of aldosterone (100-fold to 200-fold higher when plasma free levels are considered) [9••]. Epithelial aldosterone selectivity is conferred by the co-expression of MR and 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2), which converts cortisol and corticosterone into MR-inactive cortisone and 11-dehydrocorticosterone [9••]. 11βHSD2 is highly expressed in the kidney and in blood vessels but is absent in the heart [10]. Low expression levels of 11βHSD2 are observed in subcutaneous and omental adipose tissues [11]. As a consequence, GC may represent the predominant endogenous ligand in adipose tissue and perhaps in the heart. Under normal conditions, about 90% of nonepithelial MR are occupied by GC [12], but the complexes are presumably maintained inactive [13]. In the presence of oxidative stress or inflammation of the vessel wall and the heart, the GC-MR complexes become active, mimicking the deleterious effect of aldosterone in these tissues [13]. However, whether GC acts through MR in these conditions in the heart and in adipose tissue needs to be demonstrated.
Plasma Aldosterone Levels, Obesity, and Metabolic Syndrome
Aldosterone plasma levels and obesity are closely related. In 1981, Tuck et al. [14] reported for the first time that weight loss was associated with a decrease in plasma aldosterone levels. In the PAPY (Primary Aldosteronism Prevalence in Hypertension) study, Rossi et al. [15] demonstrated that plasma aldosterone concentration is independently correlated with body mass index (BMI) in overweight patients (mean BMI, 27.4 kg/m2) with hypertension. This association was also shown in normotensive, overweight adults on a high-salt diet [16]. Goodfriend et al. [17] measured visceral adipose tissue by CT, total fat mass by dual-energy x-ray absorptiometry, insulin sensitivity, and plasma aldosterone levels in 28 normotensive women and 27 normotensive men. Plasma aldosterone was directly correlated with visceral adipose tissue in women. After weight loss, plasma levels of aldosterone decreased, but the correlations between plasma aldosterone concentration and visceral adipose tissue persisted in women [17].
Increased secretion of aldosterone in the context of obesity could be related to classic determinants of aldosterone secretion such as angiotensin II, but also may be related to other stimuli such as insulin, complement C1q TNF-related protein, fatty acid oxidation products, and adipokines.
Hyperreactivity of the renin-angiotensin system (RAS) has been demonstrated in obese patients. Clinical studies showed that weight loss is associated with a decrease in aldosterone, as stated above, as well as a decrease in plasma renin activity, angiotensinogen, and angiotensin-converting enzyme (ACE) in obese patients [14, 18]. Interestingly, adipose tissue contains the complete RAS [19], which could contribute to the systemic increase in aldosterone levels found in obese patients. Engeli et al. [18] demonstrated in 70 obese women that a 5% weight loss was associated with a reduction of angiotensinogen (27%), renin (43%), and aldosterone plasma levels (31%); a decrease in ACE activity in plasma (12%); and a decrease in angiotensinogen expression in adipose tissue (20%).
Insulin resistance and hyperinsulinemia associated with obesity and the metabolic syndrome may stimulate the secretion of aldosterone. In vitro, insulin is able to stimulate aldosterone secretion by rat zona glomerulosa cells in a dose-dependent manner [20]. In agreement with this experimental observation, clinical studies have shown a strong relationship between plasma aldosterone levels and hyperinsulinemia in obese and hypertensive patients [21, 22].
Experimentally, Jeon et al. [23•] demonstrated that complement C1q TNF–related protein (CTRP)1 could also be a part of the explanation of the link between obesity and aldosterone synthesis. The authors demonstrated that in db/db mice and in obese Zucker diabetic rats, the expression of CTRP1 was increased in adipose tissue. CTRP1 was highly expressed in the adrenal gland of Sprague–Dawley rats, especially in the zona glomerulosa [23•]. In vitro, CTRP1 stimulated the production of aldosterone in human adrenocortical carcinoma cells (H295 cells). CTRP1 enhanced the expression of the aldosterone synthase gene (CYP11B2) as well as nerve growth factor–induced clone B (NGFIB) and Nur-related factor 1 (NURR1), two transcription factors that play critical roles in stimulating CYP11B2 gene expression. In conclusion, CTRP1 could be a molecular link between obesity and aldosterone production [23•].
Goodfriend et al. [24] postulated that exogenous fatty acid oxidation products, or endogenous ones from adipocytes, could stimulate aldosterone synthesis. The authors characterized a peroxidation product of linoleic acid, 12,13-epoxy-9-oxo-10(trans)-octadecanoic acid (EKODE), and demonstrated that EKODE stimulated production of aldosterone by isolated rat zona glomerulosa cells at concentrations between 1 and 30 μM. Above 50 μM, EKODE had an inhibitory effect [24]. Interestingly, EKODE was detected in plasma samples from 24 humans (12 normotensive lean, 12 hypertensive obese), and in these individuals, concentrations of EKODE correlated with plasma aldosterone levels [25]. These studies suggest that adipocytes may release free fatty acids that could stimulate aldosterone synthesis independently of angiotensin II, after being oxidized in the liver.
Ehrhart-Bornstein et al. [26] identified potent aldosterone secretory stimulants from isolated human adipocytes. In vitro, these adipokines stimulated aldosterone secretion by adrenocortical cells. This effect was independent of angiotensin II, as it persisted in the presence of valsartan, an angiotensin receptor type 1 antagonist.
In parallel, experimental data support a pathophysiologic link between aldosterone and adipose tissue development. In adrenalectomized rats, continuous infusion of aldosterone over 12 days was associated with increased body weight, related to adipose tissue hypertrophy [27]. In vitro, Caprio et al. [28] demonstrated that aldosterone induced the expression of molecular markers of adipose conversion such as adiponectin, leptin, and resistin in 3 T3-L1 cells and 3 T3-F442A cultured in a steroid-free medium. Aldosterone also induced an increase in mRNA levels of peroxisome-activated receptor-γ (PPARγ), a key transcriptional regulator of adipogenesis. 3 T3-L1 cell differentiation was blocked by MR but not by GC receptor downregulation by siRNA [28]. This study underlined the role of MR as a proadipogenic transcription factor that may mediate aldosterone effects on the development of adipose tissue.
Aldosterone, Obesity, and Inflammation
Obesity is a pro-inflammatory state characterized by systemic and adipose tissue inflammation. Obesity is associated with increased production of pro-inflammatory cytokines such as tumor necrosis factor α (TNF-α), monocyte chemoattractant protein (MCP-1), IL-6, and a decreased expression of adiponectin and PPARγ, as well as an increased level of macrophage infiltration of adipose tissue [29]. Systemic markers of inflammation such as C-reactive protein (CRP), IL-6, PAI-1, P-selectin, and vascular cell adhesion molecule 1 (VCAM-1) are associated with BMI in a large number of studies (covered in detail elsewhere [29]). In a recent ancillary study of the PREVEND (Prevention of Renal and Vascular End-Stage Disease) study, Abbasi et al. [30] demonstrated that plasma procalcitonin, a biomarker of inflammation, was associated with BMI, metabolic syndrome, and insulin resistance in the general population.
A growing body of evidence has demonstrated that activation of MR contributes to the inflammatory state associated with obesity. Guo et al. [31] showed that obese, diabetic db/db mice had an increased gene expression of proinflammatory cytokines, TNF-α, MCP-1, and pro-thrombotic factor PAI-1, and decreased expression of insulin-sensitizing factor, PPARγ, in retroperitoneal adipose tissue, compared with lean, wild-type mice. Treatment with the MR antagonist eplerenone prevented these changes in gene expression. In addition, the authors demonstrated in vitro that 3 T3-L1 cells treated with aldosterone exhibited increased expression of TNF-α and MCP-1 and reduced expression of PPARγ [31]. In ob/ob and db/db, obese mice, Hirata et al. [32] demonstrated that treatment with eplerenone significantly reduced macrophage infiltration and reactive oxygen species production in adipose tissue. Taken together, these results suggest that MR activation contributes to changes in adipose tissue that promote low-grade inflammation.
No clinical study has been conducted in obese patients to evaluate the effect of MR antagonists on inflammation. In patients with type 2 diabetes complicated by nephropathy, MR blockade reduced inflammatory markers [33]. In hypertensive patients, our group has demonstrated that treating hypertensive patients with eplerenone, compared with an atenolol regimen, resulted in reduction of pro-inflammatory mediators such as MCP-1, osteopontin, basic fibroblast growth factor (bFGF), and IL-8, in addition to reduced stiffness of subcutaneous small resistance arteries [34•].
Aldosterone and Insulin Resistance
An association between glucose intolerance and aldosterone level in patients with primary aldosteronism (PA) was demonstrated 45 years ago [35]. More recently, in a cohort of 85 patients with PA and 381 patients with essential hypertension, Fallo et al. [36] demonstrated that patients with PA had a significantly higher prevalence of metabolic syndrome (defined by ATP III criteria) than patients with essential hypertension (41.1% vs 29.6%, P < 0.005). Within the individual components of metabolic syndrome, the prevalence of impaired fasting glucose was particularly elevated in patients with PA compared with patients having essential hypertension (27% vs 15.2%, P < 0.005) [36]. The relationship between insulin resistance and elevated plasma aldosterone level is strengthened by the observation that surgical or pharmacologic treatment of PA improves insulin sensitivity [37]. The association between aldosterone level and insulin resistance has been confirmed in several populations of patients. In 356 essential hypertension patients compared with 102 normotensive patients, a significant association between plasma aldosterone concentration and insulin resistance has been reported [21]. In 302 patients with class II–IV heart failure included in the ALOFT (Aliskiren Observation of Heart Failure Treatment) study, a positive correlation between fasting insulin and plasma and urinary aldosterone levels was also demonstrated (r = 0.22, P < 0.01, and r = 0.19, P < 0.03, respectively) [38]. When patients with aldosterone escape (20%) and high urinary aldosterone excretion (34%) were compared with the rest of the studied population, early-morning fasting insulin, homeostasis model assessment of insulin resistance (HOMA-IR), and insulin/glucose ratio (IGR) were higher in the first group of patients [38]. In 84 healthy patients, Garg et al. [39] showed an independent negative correlation between the insulin sensitivity index, calculated after a 75-gram oral glucose load, and stimulated aldosterone, measured after 45 min of angiotensin II infusion (3 ng/kg per minute).
Urbanet et al. [8•] directly studied insulin sensitivity in adipose tissue from patients with PA and healthy patients who requested abdominoplasty. In vitro, in cultured adipocytes, basal and insulin-stimulated glucose uptake was unaffected by physiological concentrations of aldosterone (1–100 nM) but was impaired by pharmacologic levels (10 μM). This effect was not prevented by eplerenone, but rather by RU486, suggesting that aldosterone acts in this case, in vitro, through GR activation [8•]. Unfortunately, in this study, insulin sensitivity was tested in adipocyte cultures without differentiating healthy controls from patients with PA.
In conclusion, impaired glucose metabolism as a result of insulin resistance appears to be one of the important links between aldosterone levels and metabolic syndrome. In fact, a strong relation was shown between genetic variants of the CYP11B2 gene, which encodes for aldosterone synthase, and glucose plasma levels, both fasting and after an oral glucose challenge [40].
Mechanisms of Aldosterone-Induced Insulin Resistance
Aldosterone can impact insulin action through different mechanisms, which include oxidative stress, inflammation, and modulation of expression of insulin-signaling proteins.
Oxidative Stress and Inflammation
Kraus et al. [41] demonstrated a direct effect of aldosterone on murine brown fat tissue. Aldosterone impaired insulin-induced glucose uptake by about 25% in a dose-dependent manner, and induced the expression of the proinflammatory adipokine leptin and of MCP-1 [41].
In two models of obese mice (ob/ob and db/db), Hirata et al. [32] demonstrated that treatment with eplerenone, an MR antagonist, reduced the high levels of glucose, HOMA-IR, and plasma triglyceride concentration, and increased adiponectin levels. Reductions of macrophage infiltration, defined as F4/80 positive cells in visceral fat sections, and reactive oxygen species production were also observed after treatment of obese mice with eplerenone. The effect of aldosterone on oxidative stress was confirmed in vitro using 3 T3-L1 adipocytes treated with aldosterone. The increased intracellular levels of thiobarbituric reactive acid substances (TBARS) and greater expression of reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits p22 and p47 were decreased by pretreatment of 3 T3-L1 cells with eplerenone, and by transfection of MR-siRNAs. These results are in agreement with an MR-mediated effect on obesity-related insulin resistance partly through induction of oxidative stress and inflammation [32].
In a transgenic model of renin-angiotensin-aldosterone system (RAAS) activation and insulin resistance, the Ren2 transgenic rat, Lastra et al. [42] extended these observations to nonadipose tissues such as skeletal muscle. The authors demonstrated that treatment with spironolactone, an MR antagonist, improved systemic insulin sensitivity as evaluated by an intraperitoneal glucose tolerance test and skeletal soleal muscle glucose uptake in the presence of insulin in Ren2 rats. These effects were associated with a decrease of NADPH oxidase activity; decreased expression of the NADPH subunits NOX2, p22phox, p47phox; and a decrease in membrane lipid peroxidation in the soleus muscle of Ren 2 rats compared with Sprague–Dawley control rats. In agreement with the reduction of glucose uptake in this model, expression of insulin signaling proteins such as IRS-1, Akt, and GLUT4 was reduced in the soleus muscle of Ren2 rats. In conclusion, in this model, insulin resistance is partly mediated via MR signaling through increased oxidative stress, and is due to reduced expression of proteins involved in insulin signaling [42].
Effects on the Insulin Signaling Pathway
Wada et al. [43] demonstrated that direct exposure of 3 T3-L1 adipocytes to aldosterone for 16 h slightly reduced the amount of insulin receptor and markedly decreased the expression of insulin receptor substrate (IRS) 1 and 2 in a dose-dependent manner, as well as insulin-induced phosphorylation of Akt1, Akt2, and p44/42 mitogen-activated protein kinase (MAPK). Treatment with aldosterone for 16 h reduced insulin-induced glucose uptake in a dose-dependent manner. The amount of glucose transporters Glut 1 and Glut 4 was not affected by exposure of 3 T3-L1 adipocytes to aldosterone. These effects were prevented by pretreatment with RU486, a selective GR antagonist, and N-acetylcysteine, but not by eplerenone. Wada et al. [43] also demonstrated that aldosterone induced phosphorylation of IRS1 at Ser307 that is inhibited by rapamycin (a mammalian target of rapamycin [mTOR] pathway inhibitor) and BMS345541 (an IκB kinase β [IKKβ] inhibitor), but not by SP600125 (a c-Jun N-terminal kinase [c-JNK] inhibitor), nor by calphostin C (a protein kinase C [PKC] inhibitor). Proteasome inhibitors (lactacystin and MG132) prevented aldosterone-induced degradation of IRS1 and IRS2, but had no effect on glucose uptake. In conclusion, the authors demonstrated that aldosterone induces the degradation of IRS1 and IRS2 via GR-mediated production of reactive oxygen species and activation of IKKβ and mTOR [43].
In other cell types, such as vascular smooth muscle cells, aldosterone exerts comparable effects on IRS1. Hitomi et al. [44] demonstrated in primary culture of vascular smooth muscle cells from rats that aldosterone suppressed insulin signaling through the degradation of IRS1. This effect was inhibited by eplerenone, N-acetylcysteine, and the c-Src inhibitor PP1. The degradation of IRS1 was prevented by pretreatment with MG132, a proteasome inhibitor. In conclusion, in vascular smooth muscle cells, aldosterone decreases IRS1 expression via an MR-dependent pathway through the generation of oxidative stress.
Aldosterone Effect on Hepatic Function
Aldosterone also has an impact on other components of the regulation of glucose metabolism, such as hepatic gluconeogenesis. Aldosterone application in vitro to mouse primary cultured hepatic cells was associated with increased gene expression of glucose-6-phosphatase (G6Pase), fructose-1,6-biphosphatase, and phosphoenolpyruvate carboxykinase in a dose-dependent manner. The increased gene expression of G6Pase was not suppressed by pretreatment with spironolactone (1 μM) but was suppressed by RU-38486 (10 μM), suggesting that the effect of aldosterone on G6Pase expression was mediated through its interaction with GR [45].
In vivo, in a murine model of a high-fat, high-fructose diet (HFFD) characterized by elevated blood pressure, dyslipidemia, impaired glucose tolerance, and nonalcoholic steatohepatitis (NASH), Wada et al. [46•] recently demonstrated that treatment with spironolactone improved serum fasting glucose concentration, insulin levels, and HOMA index. The glucose area under the curve (AUC) decreased by 17.9% in the glucose tolerance test and by 33.7% in the insulin tolerance test in HFFD mice receiving spironolactone compared with untreated HFFD mice. The HFFD mice had a significantly higher concentration of glucose at any time point during the pyruvate tolerance test; this was improved by treatment with spironolactone. Thus production of hepatic glucose is increased in HFFD mice, and there is upregulation of hepatic-mRNA expression of proinflammatory cytokines such as TNF-α, IL-6, and MCP-1, and the gluconeogenic enzyme phosphoenolpyruvate carboxykinase. Treatment with spironolactone reduced expression of all of these genes in HFFD mice. This study demonstrated another level of action of aldosterone in the metabolic syndrome, involving hepatic gluconeogenesis [46•].
Aldosterone, Metabolic Syndrome, and Lipid Metabolism
Clinical studies have provided ambiguous results regarding the relationship between aldosterone levels and lipid metabolism. In 30 patients with metabolic syndrome, Goodfriend et al. [22] demonstrated a strong negative correlation between plasma aldosterone level and HDL cholesterol level. The patients with the lowest HDL had the highest BMI, so obesity could drive the observed association between aldosterone and HDL cholesterol [22]. This association was confirmed in another study conducted in 356 people in the Seychelles, and the association persisted after adjustment for BMI [47]. However, in 2,891 participants in the Framingham Offspring Study, plasma aldosterone levels were not independently correlated with HDL cholesterol [48]. Small interventional studies have yielded contradictory results regarding the effects of MR antagonists on lipid metabolism. In 16 patients with type 2 diabetes, Joffe et al. [49] described a significant reduction in triglyceride levels after 6 weeks of treatment with eplerenone, without any change in total or HDL cholesterol concentrations. In contrast, a study by Matsumoto et al. [50], conducted in 33 patients with type 2 diabetes complicated by diabetic nephropathy, did not show similar results.
In a rodent model of metabolic syndrome induced by HFFD and in obese mice, treatment with spironolactone was associated with decreases in triglycerides and total cholesterol [46•]. This finding was confirmed for circulating triglycerides in other studies involving obese mice treated with eplerenone [31, 32]. However, in SHR/NDmcr-cps rats (a rat model of metabolic syndrome), no significant reduction of triglyceride levels could be found with eplerenone administration [51].
Aldosterone, the Cardiovascular System, and Metabolic Syndrome
Increased plasma aldosterone levels observed in the metabolic syndrome could affect cardiovascular structures and the kidney, and may participate in the increased cardiovascular risk of these patients. Two large clinical trials, EPHESUS (Eplerenone Post–Acute Myocardial Infarction Heart Failure Efficacy and Survival Study) [52] and RALES (Randomized Aldactone Evaluation Study, using spironolactone in heart failure patients) [53], confirmed the beneficial effect of adding MR antagonists to standard therapy including ACE inhibitors and angiotensin II receptor blockers; overall mortality, rate of cardiovascular hospitalization, and cardiovascular death were reduced, compared with rates for patients who received standard medical therapy. Clinical trials are still needed to test the efficacy of MR antagonists in reducing cardiovascular risk in metabolic syndrome patients without heart failure.
In a high-salt environment, aldosterone induces inflammation and oxidative stress in the vascular wall, in the heart and the kidney. These in turn lead to cardiovascular fibrosis and hypertrophy, glomerulosclerosis, tubulointerstitial fibrosis, and podocyte dysfunction [5]. It has been clearly demonstrated in the past that administering mineralocorticoid and salt to uninephrectomized rats [54, 55] or Wistar rats [56] induces severe vascular and cardiac hypertrophy and fibrosis. These effects were prevented by spironolactone, an MR antagonist, independently of blood pressure [56]. In addition, MR antagonist treatment of stroke-prone spontaneously hypertensive rats (SHR) prevented cardiac and vascular remodeling and endothelial dysfunction [57]. Oxidative stress and inflammation play a central role in the deleterious effects of aldosterone and MR signaling on the cardiovascular system. Increased oxidative stress and inflammation in the heart and vessels has been reported in rats treated with deoxycorticosterone acetate (DOCA) and salt [58, 59] or aldosterone and salt [56]. Vascular inflammation and oxidative stress precede the development of cardiac and vascular fibrosis [60]. Treating rats that are hypertensive due to aldosterone and salt with apocynin, an NADPH oxidase inhibitor [57], prevented the cardiovascular remodeling and fibrosis; the same result occurred in uninephrectomized aldosterone/salt hypertensive rats treated with pyrrolidine dithiocarbamate (PDTC) or N-acetylcysteine [60]. In a DOCA/salt hypertensive rat model, our group demonstrated that administering an endothelin receptor type A (ETAR) antagonist reduced cardiac collagen deposition [61]. In addition, in aldosterone/salt hypertensive rats, an ETAR antagonist prevented cardiovascular fibrosis and vascular remodeling [62] and reduced oxidative stress and expression of intercellular adhesion molecule-1 (ICAM-1) in the vascular wall [63]. These studies emphasize the role of an activated endothelin system in mineralocorticoid-induced cardiovascular damage. Effects on the kidney were demonstrated by Greene et al. [64], who showed that aldosterone induced renal injury independently of angiotensin II and demonstrated that the antiproteinuric effect of angiotensin II blockade in the remnant-kidney rat were reversed by aldosterone infusion. In addition, administering aldosterone and salt to normotensive rats [65] and to uninephrectomized rats [66] induced renal injury characterized by proteinuria, mesangial matrix expansion and cell proliferation, glomerulosclerosis, tubulointerstitial inflammation, and podocyte changes. Increased oxidative stress markers and inflammation were also described in the kidney in these models [65, 66]. Indeed, treatment with Tempol, an antioxidant, prevented kidney damage induced by aldosterone and salt [65, 66]. Taken together, these studies emphasized the deleterious effects of aldosterone on the heart, the vessels, and the kidney through induction of oxidative stress and inflammation.
The increased level of aldosterone associated with metabolic syndrome could play a role in cardiac and renal disease. The rat model of metabolic syndrome, SHR/NDmcr-cp (SHR/cp), is characterized by hypertension (derived from a background of SHR) and obesity due to a nonsense mutation in the leptin receptor gene. In this model, metabolic abnormalities include hyperinsulinemia, dyslipidemia, and hypertriglyceridemia, all of which are consistent with metabolic syndrome [67]. Gross et al. [68] demonstrated that, compared with streptozotocin-treated rats, SHR/cp exhibited marked structural kidney lesions such as podocyte damage and mesangial matrix expansion. Circulating aldosterone levels and glomerular expression of serum and glucocorticoid-inducible kinase 1 (Sgk1), reflecting MR signaling, were increased in SHR/cp rats compared to SHR. Treatment of SHR/cp rats with eplerenone and Tempol, an antioxidant, improved podocyte injury and proteinuria and decreased urinary 8-hydroxy-2′-deoxyguanosine (8-OHdG) and expression of NADPH subunits. Taken together, these results show that in the metabolic syndrome, aldosterone signaling contributes to podocyte injury via oxidative stress [67].
Salt load plays a critical role in cardiovascular and renal injury induced by aldosterone and/or MR signaling in the metabolic syndrome. In SHR/cp rats, Matsui et al. [51] demonstrated that left ventricular diastolic dysfunction was observed only in salt-loaded conditions. Salt-load SHR/cp also exhibited severe hypertension, perivascular fibrosis, overproduction of reactive oxygen species, and upregulation of Sgk-1 (reflecting MR signaling in the heart), compared with SHR and SHR/cp on a normal-salt diet. Administration of eplerenone or the antioxidant superoxide dismutase mimetic Tempol prevented a high-salt diet from inducing left ventricular diastolic dysfunction and oxidative stress in SHR/cp. These results suggest that SHR/cp do not exhibit cardiovascular dysfunction or oxidative stress and fibrosis with a normal-salt diet. However, the metabolic syndrome in this rat model is a predisposing condition for salt-induced cardiac dysfunction and vascular fibrosis via MR signalling, possibly through the increased levels of oxidative stress [51]. In the kidney, a salt load induced advanced glomerulosclerosis and tubulointerstitial fibrosis, podocyte damage, inflammation, and oxidative stress in SHR/cp, compared with SHR/cp receiving a normal-salt diet and SHR on a high-salt diet. Tempol and eplerenone significantly improved these abnormalities. Tempol reduced the expression of renal Sgk1, which may suggest that oxidative stress plays a role in MR activation in this model [69].
Conclusions
The metabolic syndrome and obesity are associated with increased plasma aldosterone levels. Aldosterone, in turn, plays a role in adipocyte function, local and systemic inflammation, and insulin resistance. High concentrations of aldosterone could also contribute to cardiovascular and renal injury, especially in a high-salt environment, and to the increased overall cardiovascular risk of these patients. However, a large interventional trial testing the efficacy of MR antagonists in reducing the cardiovascular risk of patients with metabolic syndrome is needed to confirm these experimental observations.
References
Recently published papers of interest have been highlighted as: • Of importance •• Of major importance
Eckel RH, Alberti KG, Grundy SM, Zimmet PZ: The metabolic syndrome. Lancet 2010, 375:181–183.
•• Alberti KG, Eckel RH, Grundy SM, et al.: Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120:1640–1645. This paper is of particular interest, as it describes the new diagnostic criteria for the metabolic syndrome.
Lakka HM, Laaksonen DE, Lakka TA, et al.: The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA 2002, 288:2709–2716.
• Motillo S, Filion KB, Genest J, et al.: The metabolic syndrome and cardiovascular risk: a systematic review and meta-analysis. J Am Coll Cardiol 2010, 56:1113–1132. This recent meta-analysis described the high cardiovascular risk associated with the metabolic syndrome, using the definition of the National Cholesterol Education Program.
Briet M, Schiffrin EL: Aldosterone: effects on the kidney and cardiovascular system. Nat.Rev.Nephrol. 2010, 6:261–273.
Ingelsson E, Pencina MJ, Tofler GH, et al.: Multimarker approach to evaluate the incidence of the metabolic syndrome and longitudinal changes in metabolic risk factors: the Framingham Offspring Study. Circulation 2007, 116:984–992.
Chartier L, Schiffrin E, Thibault G, et al.: Atrial natriuretic factor inhibits the stimulation of aldosterone secretion by angiotensin II, ACTH and potassium in vitro and angiotensin II-induced steroidogenesis in vivo. Endocrinology 1984, 115:2026–2028.
• Urbanet R, Pilon C, Calcagno A, et al.: Analysis of insulin sensitivity in adipose tissue of patients with primary aldosteronism. J Clin Endocrinol Metab 2010, 95:4037–4042. This study is the first one to test insulin sensitivity in adipose tissue obtained from patients with primary aldosteronism and controls. The authors did not demonstrate insulin resistance in vitro after aldosterone administration in primary adipocyte cell culture.
•• Funder JW: Reconsidering the roles of the mineralocorticoid receptor. Hypertension 2009, 53:286–290. This review raises the important question of the role of glucocorticoids acting through MR activation.
Edwards CR, Stewart PM, Burt D, et al.: Localisation of 11 beta-hydroxysteroid dehydrogenase—tissue specific protector of the mineralocorticoid receptor. Lancet 1988, 2:986–989.
Veilleux A, Laberge PY, Morency J, et al.: Expression of genes related to glucocorticoid action in human subcutaneous and omental adipose tissue. J Steroid Biochem Mol Biol 2010, 122:28–34.
Funder J, Myles K: Exclusion of corticosterone from epithelial mineralocorticoid receptors is insufficient for selectivity of aldosterone action: in vivo binding studies. Endocrinology 1996, 137:5264–5268.
Funder JW, Reincke M: Aldosterone: a cardiovascular risk factor? Biochim Biophys Acta 2010, 1802:1188–1192.
Tuck ML, Sowers J, Dornfeld L, et al.: The effect of weight reduction on blood pressure, plasma renin activity, and plasma aldosterone levels in obese patients. N Engl J Med 1981, 304:930–933.
Rossi GP, Belfiore A, Bernini G, et al.: Body mass index predicts plasma aldosterone concentrations in overweight-obese primary hypertensive patients. J Clin Endocrinol Metab 2008, 93:2566–2571.
Bentley-Lewis R, Adler GK, Perlstein T, et al.: Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J Clin Endocrinol Metab 2007, 92:4472–4475.
Goodfriend TL, Kelley DE, Goodpaster BH, Winters SJ: Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res 1999, 7:355–362.
Engeli S, Bohnke J, Gorzelniak K, et al.: Weight loss and the renin-angiotensin-aldosterone system. Hypertension 2005, 45:356–362.
Gorzelniak K, Engeli S, Janke J, et al.: Hormonal regulation of the human adipose-tissue renin-angiotensin system: relationship to obesity and hypertension. J Hypertens 2002, 20:965–973.
Petrasek D, Jensen G, Tuck M, Stern N: In vitro effects of insulin on aldosterone production in rat zona glomerulosa cells. Life Sci 1992, 50:1781–1787.
Colussi G, Catena C, Lapenna R, et al.: Insulin resistance and hyperinsulinemia are related to plasma aldosterone levels in hypertensive patients. Diabetes Care 2007, 30:2349–2354.
Goodfriend TL, Egan B, Stepniakowski K, Ball DL: Relationships among plasma aldosterone, high-density lipoprotein cholesterol, and insulin in humans. Hypertension 1995, 25:30–36.
• Jeon JH, Kim KY, Kim JH, et al.: A novel adipokine CTRP1 stimulates aldosterone production. FASEB J 2008, 22:1502–1511. This paper provides one of the explanations for the link between obesity and aldosterone synthesis. The authors demonstrated that C1q TNF-related protein (CTRP)1, an adipokine, stimulates the synthesis of aldosterone by cells from the human adrenal cortical cell line H295R.
Goodfriend TL, Ball DL, Raff H, et al.: Oxidized products of linoleic acid stimulate adrenal steroidogenesis. Endocr Res 2002, 28:325–330.
Goodfriend TL, Ball DL, Egan BM, et al.: Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension 2004, 43:358–363.
Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A, et al.: Human adipocytes secrete mineralocorticoid-releasing factors. Proc Natl Acad Sci U S A 2003, 100:14211–14216.
Devenport LD, Goodwin KG, Hopkins PM: Continuous infusion of aldosterone: correlates of body weight gain. Pharmacol Biochem Behav 1985, 22:707–709.
Caprio M, Feve B, Claes A, et al.: Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J 2007, 21:2185–2194.
Berg AH, Scherer PE: Adipose tissue, inflammation, and cardiovascular disease. Circ Res 2005, 96:939–949.
Abbasi A, Corpeleijn E, Postmus D, et al.: Plasma procalcitonin is associated with obesity, insulin resistance, and the metabolic syndrome. J Clin Endocrinol Metab 2010, 95:E26–E31.
Guo C, Ricchiuti V, Lian BQ, et al.: Mineralocorticoid receptor blockade reverses obesity-related changes in expression of adiponectin, peroxisome proliferator-activated receptor-gamma, and proinflammatory adipokines. Circulation 2008, 117:2253–2261.
Hirata A, Maeda N, Hiuge A, et al.: Blockade of mineralocorticoid receptor reverses adipocyte dysfunction and insulin resistance in obese mice. Cardiovasc Res 2009, 84:164–172.
Takebayashi K, Matsumoto S, Aso Y, Inukai T: Aldosterone blockade attenuates urinary monocyte chemoattractant protein-1 and oxidative stress in patients with type 2 diabetes complicated by diabetic nephropathy. J Clin Endocrinol Metab 2006, 91:2214–2217.
• Savoia C, Touyz RM, Amiri F, Schiffrin EL: Selective mineralocorticoid receptor blocker eplerenone reduces resistance artery stiffness in hypertensive patients. Hypertension 2008, 51:432–439. This clinical study involving hypertensive patients demonstrated the beneficial effect of eplerenone, an MR blocker, on inflammation and resistance artery stiffness.
Conn JW: Hypertension, the potassium ion and impaired carbohydrate tolerance. N Engl J Med 1965, 273:1135–1143.
Fallo F, Veglio F, Bertello C, et al.: Prevalence and characteristics of the metabolic syndrome in primary aldosteronism. J Clin Endocrinol Metab 2006, 91:454–459.
Catena C, Lapenna R, Baroselli S, et al.: Insulin sensitivity in patients with primary aldosteronism: a follow-up study. J Clin Endocrinol Metab 2006, 91:3457–3463.
Freel EM, Tsorlalis IK, Lewsey JD, et al.: Aldosterone status associated with insulin resistance in patients with heart failure—data from the ALOFT study. Heart 2009, 95:1920–1924.
Garg R, Hurwitz S, Williams GH, et al.: Aldosterone production and insulin resistance in healthy adults. J Clin Endocrinol Metab 2010, 95:1986–1990.
Ranade K, Wu KD, Risch N, et al.: Genetic variation in aldosterone synthase predicts plasma glucose levels. Proc Natl Acad Sci U S A 2001, 98:13219–13224.
Kraus D, Jager J, Meier B, et al.: Aldosterone inhibits uncoupling protein-1, induces insulin resistance, and stimulates proinflammatory adipokines in adipocytes. Horm Metab Res 2005, 37:455–459.
Lastra G, Whaley-Connell A, Manrique C, et al.: Low-dose spironolactone reduces reactive oxygen species generation and improves insulin-stimulated glucose transport in skeletal muscle in the TG(mRen2)27 rat. Am J Physiol Endocrinol Metab 2008, 295:E110–E116.
Wada T, Ohshima S, Fujisawa E, et al.: Aldosterone inhibits insulin-induced glucose uptake by degradation of insulin receptor substrate (IRS) 1 and IRS2 via a reactive oxygen species-mediated pathway in 3 T3-L1 adipocytes. Endocrinology 2009, 150:1662–1669.
Hitomi H, Kiyomoto H, Nishiyama A, et al.: Aldosterone suppresses insulin signaling via the downregulation of insulin receptor substrate-1 in vascular smooth muscle cells. Hypertension 2007, 50:750–755.
Yamashita R, Kikuchi T, Mori Y, et al.: Aldosterone stimulates gene expression of hepatic gluconeogenic enzymes through the glucocorticoid receptor in a manner independent of the protein kinase B cascade. Endocr J 2004, 51:243–251.
• Wada T, Kenmochi H, Miyashita Y, et al.: Spironolactone improves glucose and lipid metabolism by ameliorating hepatic steatosis and inflammation and suppressing enhanced gluconeogenesis induced by high-fat and high-fructose diet. Endocrinology 2010, 151:2040–2049. The authors demonstrated the role of MR activation in hepatic steatosis, inflammation, and oxidative stress in mice receiving a high-fat and high-fructose diet. They also demonstrated that MR activation is implicated in hepatic gluconeogenesis, which could participate in the impairment of glucose metabolism associated with metabolic syndrome.
Bochud M, Nussberger J, Bovet P, et al.: Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension 2006, 48:239–245.
Kathiresan S, Larson MG, Benjamin EJ, et al.: Clinical and genetic correlates of serum aldosterone in the community: the Framingham Heart Study. Am J Hypertens 2005, 18:657–665.
Joffe HV, Kwong RY, Gerhard-Herman MD, et al.: Beneficial effects of eplerenone versus hydrochlorothiazide on coronary circulatory function in patients with diabetes mellitus. J Clin Endocrinol Metab 2007, 92:2552–2558.
Matsumoto S, Takebayashi K, Aso Y: The effect of spironolactone on circulating adipocytokines in patients with type 2 diabetes mellitus complicated by diabetic nephropathy. Metabolism 2006, 55:1645–1652.
Matsui H, Ando K, Kawarazaki H, et al.: Salt excess causes left ventricular diastolic dysfunction in rats with metabolic disorder. Hypertension 2008, 52:287–294.
Pitt B, Remme W, Zannad F, et al.: Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003, 348:1309–1321.
Pitt B, Zannad F, Remme WJ, et al.: The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999, 341:709–717.
Young M, Fullerton M, Dilley R, Funder J: Mineralocorticoids, hypertension, and cardiac fibrosis. J Clin Invest 1994, 93:2578–2583.
Brilla CG, Weber KT: Mineralocorticoid excess, dietary sodium, and myocardial fibrosis. J Lab Clin Med. 1992, 120:893–901.
Nakano S, Kobayashi N, Yoshida K, et al.: Cardioprotective mechanisms of spironolactone associated with the angiotensin-converting enzyme/epidermal growth factor receptor/extracellular signal-regulated kinases, NAD(P)H oxidase/lectin-like oxidized low-density lipoprotein receptor-1, and Rho-kinase pathways in aldosterone/salt-induced hypertensive rats. Hypertens Res 2005, 28:925–936.
Endemann DH, Touyz RM, Iglarz M, et al.: Eplerenone prevents salt-induced vascular remodeling and cardiac fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension 2004, 43:1252–1257.
Somers MJ, Mavromatis K, Galis ZS, Harrison DG: Vascular superoxide production and vasomotor function in hypertension induced by deoxycorticosterone acetate-salt. Circulation 2000, 101:1722–1728.
Beswick RA, Zhang H, Marable D, et al.: Long-term antioxidant administration attenuates mineralocorticoid hypertension and renal inflammatory response. Hypertension 2001, 37:781–786.
Sun Y, Zhang J, Lu L, et al.: Aldosterone-induced inflammation in the rat heart : role of oxidative stress. Am J Pathol 2002, 161:1773–1781.
Ammarguellat F, Larouche I, Schiffrin EL: Myocardial fibrosis in DOCA-salt hypertensive rats: effect of endothelin ET(A) receptor antagonism. Circulation 2001, 103:319–324.
Park JB, Schiffrin EL: Cardiac and vascular fibrosis and hypertrophy in aldosterone-infused rats: role of endothelin-1. Am J Hypertens 2002, 15:164–169.
Pu Q, Neves MF, Virdis A, et al.: Endothelin antagonism on aldosterone-induced oxidative stress and vascular remodeling. Hypertension 2003, 42:49–55.
Greene EL, Kren S, Hostetter TH: Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996, 98:1063–1068.
Nishiyama A, Yao L, Nagai Y, et al.: Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension 2004, 43:841–848.
Shibata S, Nagase M, Yoshida S, et al.: Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension 2007, 49:355–364.
Nagase M, Yoshida S, Shibata S, et al.: Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J.Am.Soc.Nephrol. 2006, 17:3438–3446.
Gross ML, Ritz E, Schoof A, et al.: Comparison of renal morphology in the Streptozotocin and the SHR/N-cp models of diabetes. Lab Invest 2004, 84:452–464.
Nagase M, Matsui H, Shibata S, et al.: Salt-induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: role of oxidative stress. Hypertension 2007, 50:877–883.
Source of Funding
The work of the authors was supported by Canadian Institutes of Health Research (CIHR) grants MOP37137, MOP82790, and MOP102606; a Canada Research Chair (CRC) on Hypertension and Vascular Research from the CIHR/CRC Program of the Government of Canada; the Canada Fund for Innovation (E.L.S.); and a Fellowship from the Heart and Stroke Foundation of Canada (M.B.).
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Briet, M., Schiffrin, E.L. The Role of Aldosterone in the Metabolic Syndrome. Curr Hypertens Rep 13, 163–172 (2011). https://doi.org/10.1007/s11906-011-0182-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-011-0182-2