
Abstract
Early in the HIV epidemic, the central nervous system (CNS) was recognized as a target of infection and injury in the advanced stages of disease. Though the most severe forms of HIV-associated neurocognitive disorder (HAND) related to severe immunosuppression are rare in the current era of widespread combination antiretroviral therapy (cART), evidence now supports pathological involvement of the CNS throughout the course of infection. Recent work suggests that the stage for HIV neuropathogenesis may be set with initial viral entry into the CNS, followed by initiation of pathogenetic processes including neuroinflammation and neurotoxicity, and establishment of local, compartmentalized HIV replication that may reflect a tissue reservoir for HIV. Key questions still exist as to when HIV establishes local infection in the CNS, which CNS cells are the primary targets of HIV, and what mechanistic processes underlie the injury to neurons that produce clinical symptoms of HAND. Advances in these areas will provide opportunities for improved treatment of patients with established HAND, prevention of neurological disease in those with early stage infection, and understanding of HIV tissue reservoirs that will aid efforts at HIV eradication.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Early in the HIV epidemic, a large proportion of the neurological manifestations of HIV presented as opportunistic central nervous system (CNS) infections, including toxoplasmosis and progressive multifocal leukoencephalopathy [1]. By 1987, the nonspecific “subacute encephalitis” that widely affected patients with HIV was identified as the AIDS dementia complex (ADC, now termed HIV-associated dementia, or HAD) and was recognized as a manifestation of HIV itself rather than that of an alternate infection [2]. Estimates suggest that 20–30 % of HIV patients with uncontrolled HIV ultimately developed HAD in the pre-combination antiretroviral therapy (cART) era [3].
With the development of cART, the incidence of HAD in persons living with HIV decreased dramatically. For patients with well-controlled HIV, long survival with relative immune preservation, often with extended periods of undetectable viral loads, has been also associated with dramatically lower prevalence of HAD [4]. Even so, neurological effects of HIV are seen in chronically infected patients and are now grouped under the broader term, HIV-associated neurocognitive disorders, or HAND [5]. In different contexts, 18–50 % of patients with chronic HIV on cART manifest HAND, ranging from asymptomatic neurocognitive impairment to fully manifest HAD [6, 7]. Though HAND still affects a large subset of the HIV-infected population, the neuropathogenesis of HIV remains a dynamic puzzle. In an effort to better understand how HIV affects the CNS, studies now encompass topics from viral entry and primary infection to mechanistic processes that underlie HIV-associated damage to the CNS as well as potential viral persistence in this tissue site. Recent critical strides have been made in the understanding of the neuropathogenesis of HIV that provide promise for improved cART treatment strategies and potentially adjunctive therapy in the context of well-controlled HIV.
Viral Entry and Establishment of Inflammation and Injury in the CNS
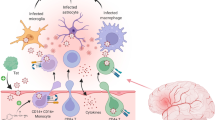
Although HIV-related pathology and the virus itself have been documented in brain tissues of patients with advanced AIDS and HAD since early in the epidemic [8, 9], ongoing studies have focused on understanding when and how HIV penetrates the blood-brain barrier (BBB) to enter the CNS and establish local infection, and on processes underlying neuropathogenesis (see Fig. 1) [10, 11]. While prior anecdotal cases suggested that HIV neuroinvasion occurs early in the progression of systemic HIV infection [12–14], recent systematic work now shows that viral entry into the CNS occurs almost immediately after systemic infection. One recent study focusing on individuals evaluated during acute HIV infection detected HIV in the CNS compartment (cerebrospinal fluid, CSF) after an estimated 8 days post initial infection [15].

Potential mechanisms of HIV-related CNS injury prior to combination antiretroviral therapy (cART). Systemic activation of immune cells stimulates increased transmigration of lymphocytes and monocytes across the blood-brain barrier (BBB). Once in the central nervous system (CNS) compartment, immune cells release pro-inflammatory signals that stimulate further immune cell influx (cytokines such as monocyte chemoattractant protein-1 and interferon-inducible protein-10) and matrix metalloproteinases that disrupt the BBB. Some of the imported cells are infected with HIV, allowing for local production of virions that enter resident CNS cells including perivascular macrophages, brain microglial cells, and astrocytes. Macrophage, microglial, and T lymphocyte infection may support local CNS HIV replication, facilitating emergence of unique “compartmentalized” CNS HIV that reflects production of HIV independent from the periphery. Activated microglia and perivascular macrophages release neurotoxic immune products that lead to neuronal dysfunction; this activation stimulates production of neopterin, a pteridine biomarker of immune activation readily measured in the cerebrospinal fluid. Potentially neurotoxic HIV proteins such as HIV Tat may freely cross the BBB or be released by resident HIV infected cells. Astrocytes harboring HIV virions or fragments may not facilitate viral replication, but may contribute to neuropathogenesis through multiple mechanisms including injury to the blood-brain barrier and release of neurotoxic products
While it seems that the virus enters the CNS almost immediately, the mechanism by which it does so remains poorly understood. Different entry mechanisms have been suggested for CNS infection, from free virus entry to cellular-mediated entry. The true infection mechanism may involve a combination of effects. The well-accepted “Trojan horse” hypothesis suggests that the virus enters mainly through CD4 T lymphocytes or possibly monocytes during routine surveillance, only to go on infecting local cells of the CNS [16, 17]. Recent findings suggest that HIV additionally disrupts the integrity of the BBB, through altering expression of and posttranslationally modifying proteins critical to maintaining BBB endothelial tight junctions [18–20]. Interestingly, Tat, a protein implicated in these modifications, shows differential effects in vitro on BBB elements in HIV subtype B and recombinant CRF02_AG [21]. Thus, while research on specific clades is instructive, the diversity of HIV suggests a diversity of entry mechanisms. Further evidence in support of the concept that HIV is carried into the CNS via immune cells rather than as free virus is found in studies employing natalizumab, a blocker of alpha-four integrin that prevents passage of both lymphocytes and monocytes across the endothelial cells comprising the blood-brain barrier [22]. When administered to rhesus macaques during acute SIV infection, natalizumab was associated with profoundly reduced levels of HIV DNA in the brain tissue of sacrificed animals as compared with untreated animals. These studies did not utilize antiretroviral treatment, so the levels of cell-free HIV RNA were presumably high in the blood, and not prevented by use of natalizumab, suggesting that the almost undetectable levels of HIV DNA in the brain were due to reductions in virus trafficking by immune cells.
After viral entry during acute infection, inflammation and immune activation in the brain and CSF compartments can be detected immediately [15, 23, 24]. This initial burst of inflammatory perturbation appears to be a trigger which initiates a cascade of immune activation within the CNS. Recent studies indicate that levels of inflammatory metabolites measured by brain magnetic resonance spectroscopy (MRS) and soluble and cellular markers of immune activation in the CSF progressively increase during the first months to years of untreated infection [25, 26•], likely providing the basis of neurological injury and persistent infection in later stage disease.
CNS Compartmentalization
While HIV seems to gain early access to the brain, it is unclear whether one entry event seeds the CNS with virus, or whether there are multiple entry points throughout the course of infection, and whether discrete breaches or a general vulnerability of the BBB contribute to CNS HIV infection. The localized infection of HIV in the CNS and the subsequent genetic changes found in HIV strains identified in CSF and brain tissue have been termed “CNS compartmentalization” of HIV (Fig. 1). As quantitative and computational genetics methods improve, the analysis of different genetic variants may have the potential to offer insights about the timeline of establishment of local CNS infection and thus the existence of a discrete “reservoir” for HIV replication within the CNS.
Previous research has shown that HIV replication can occur in the CNS, offering the virus a potential mechanism of continuity in and adaptation to the distinct CNS environment as early as during the primary HIV infection [27]. CD8+ T cells may control CNS SIV viral load; the depletion of CD8+ T cells in the CNS has previously been shown to increase the viral load [28, 29], suggesting that a CNS source of HIV replication is controlled or modulated by CD8+ T cells. Whether ongoing seeding from the periphery or local CNS replication in cells of the CNS is the source of HIV detected in the CSF and brain is not entirely known and may differ in distinct stages of infection and in cART-naive versus cART-suppressed contexts. Using computational methods in an SIV model, the Salemi group has recently suggested that continual CNS viral seeding remains an important factor in persistent CNS HIV infection in cART-naïve animals, with the final event stemming from virus in the bone marrow [30]. Extensive studies of CNS compartmentalization have revealed not only distinct viral variants in CNS versus blood [31–33] but also evidence of compartmentalized viral evolution of local CNS HIV envelope sequences in the setting of viral replication unchecked by cART [34]. In an initial study examining HIV during suppressive ART in the CSF and plasma, no evidence was found suggesting that continual viral replication in the CNS was the major factor allowing viral persistence in the CNS on ART, though this study was small and further investigations in this area are needed [35•].
The CNS as a Potential Reservoir
While HIV reservoirs in the gut and bone marrow have been confirmed, the concept of a CNS reservoir for HIV has recently gained attention and support. This new attention to the CNS as a potential reservoir arises in part from studies in macaque models of CNS HIV with accelerated rates of HIV encephalitis that demonstrate HIV DNA in the brain prior to and in the presence of cART [36–38]. More recently, human studies of typical HIV infection have indicated localized CNS evolution of HIV prior to ART [34] and, importantly, have demonstrated that HIV can independently replicate in the CNS despite systemically suppressive ART (commonly termed “CSF escape”) [39••, 40]. CNS macrophages and microglial cells have been historically considered the most likely sources of sustained HIV replication in the CNS because of their long-lived nature and the substantial evidence that in macaques and humans, advanced forms of HIV encephalitis and even presymptomatic disease involve infection of these cell types [36, 41, 42, 43••]. Specific antiretrovirals have a distinct ability to enter and prevent retroviral replication in myeloid lineage cells [44], which may provide a substrate of persistent HIV replication in the CNS in the face of inadequate viral suppression in this compartment within this particular cell type. Populations of resident CNS macrophage and microglial cells may be established during brain development rather than being replenished by circulating monocytes [45, 46]. The fact that microglial cells do not turn over in stem cell transplantation without brain irradiation [47] is a potential limitation of this procedure in achievement of total body HIV eradication and could be posited to contribute to “failure” of this approach in recently reported cases [48].
Evidence has suggested differential viral decay rates for CNS-derived HIV after initiation of cART [49], suggesting that HIV may replicate within distinct cell types within the CNS. Using assays to assess HIV’s ability to enter and replicate in cells with distinct cellular surface markers, Swanstrom and colleagues have noted two distinct patterns of cellular tropism in CSF HIV derived from patients with unique, compartmentalized variants that are presumed to derive from the CNS. While some CNS HIV were able to replicate in cells with low numbers of CD4 receptors, consistent with macrophage/microglial cells, other compartmentalized viruses were dependent on high levels of CD4 receptors, suggesting adaptation to replication within T cells [34]. Unlike activated macrophages and CD8+ T cells, CD4+ T cells have not been classically thought to be present in large concentrations in the SIV- or HIV-infected brain, but the suggestion that unique viral variants detected in the CNS may derive from T cells raises the question of alternate T cell reservoirs in the CNS compartment, potentially resident in the brain, choroid plexus, or lymphoid follicles in the meninges.
Additionally, based on studies employing basic histopathology as well as laser capture microdissection [9, 41, 50], astrocytes have emerged as potential host cells for HIV. However, as astrocytes lack CD4 receptors, the mechanism of entry into astrocytes has been a mystery. Now, evidence in a SIV model suggests that CNS compartmentalization may create selective pressures that allow the virus to enter cells independently of CD4 receptors, allowing the virus to enter astrocytes [51•]. Inhibition of a specific receptor decreased replication and resulted in a decreased latent reservoir in the SIV model, suggesting the receptor might be important in the development of this particular CNS HIV reservoir. Initial studies have suggested that while HIV proteins or nucleic acid may be found in astrocytes, HIV does not seem to replicate in these cells and that full-HIV genomes are not detected [50]. However, the presence of HIV proteins and fragments in astrocytes has been associated with astrocyte activation, dysfunction, and blood-brain barrier disruption, likely a key element of CNS pathogenesis [52].
Future studies are critical to understand the distribution of HIV and its potential persistence in the CNS, including where the virus is actively replicating prior to cART and whether latent or even replicating HIV is detected in more than rare “CSF escape” individuals in systemically suppressive cART.
CNS Injury: Neurotoxicity or Neuroinflammation, or Both?
Since the effects of HAND include cognitive impairment, HIV must by necessity either directly or indirectly contribute to neuronal dysfunction or damage. HIV-related CNS damage may begin very soon after infection; elevations of neurofilament light chain (NFL), a CSF biomarker associated with neuronal injury [53], and reductions of magnetic resonance spectroscopy markers of neuronal function [54] have been documented in individuals studied within the first year of infection. Key pathogenic models of neuroimmune injury are predicated upon enhanced trafficking of immune cells and soluble pro-immune molecules to the CNS, release of cytokines and chemokines that amplify this cycle of neuroinflammation, and production of neurotoxic molecules which damage neurons (reviewed in [3, 17, 55]). Recent investigations highlight specific mechanisms, ranging from the impact of the oxidative stress of inflammatory responses, to HIV protein Tat’s interference with neuronal transcriptional pathways.
In advanced infection, CNS immune activation, such as monocyte activation, has been associated with neurocognitive impairment [56, 57]. A recent clinical study confirmed that in ART-naïve patients with advanced HIV, those with HAND exhibited higher levels of inflammatory chemokines and cytokines than those who did not exhibit HAND, but had comparable HIV RNA levels [58]. New evidence points specifically to oxidative stress associated with the inflammatory response, suggesting that this is damaging to neuronal processes and other brain cells. Heme-oxygenase-1, a detoxifying enzyme, was recently found to be deficient in the brains of individuals with HAND [59]. Infection in the CNS generates persistent inflammation even in patients with well-controlled HIV [60, 61], but it is still not known whether CNS immune activation is sufficient for progression of HAND.
One protein that has garnered attention for its potentially widespread effects in CNS HIV is the tat gene-encoded HIV protein Tat. Tat is a transactivating protein that influences the expression of both host and viral gene expression; its functions have been reviewed elsewhere [62]. Tat has been shown to be the target of CD4+ T cell autophagy, highlighting a potentially critical role for the transactivator in the neuropathogenesis of HIV [63]. Though prior studies have not clearly shown an association between Tat concentrations or activity and human neurological or cognitive disorders, recent findings that Tat can induce neurobehavioral changes independent of inflammatory response in a neonatal rodent model have highlighted the potential clinical importance of this protein [64]. It is unclear whether these changes are completely identical to HAND or whether neuroinflammation worsens the effect of these changes. Nevertheless, this study suggests that neuroinflammation may not be the sole factor in neurodegeneration. Lending credence to this concept, in an SIV model, some neuroprotection was achieved using a combination experimental therapy, despite continued virus replication and neuroinflammation [65•].
Neurotoxic astrocyte activity may also be provoked through Tat’s induction of PZX7R, a receptor involved in generating an inflammatory response in the brain [66] that upregulates platelet-derived growth factor (PDGF). PDGF induces MCP-1 production by astrocytes and in turn increases monocyte migration across the blood-brain barrier, suggesting a role for astrocytes in the proinflammatory CNS response [67]. Infected astrocytes have also been implicated in directly inducing apoptosis in surrounding cells, by deregulation of cytochrome C processes [68•]. Previous work suggests that another possible mechanism of neuron loss is excitotoxicity mediated by NMDA receptors, causing loss of synapses. In one study, this effect was reversed by NMDA antagonists, sparing synapses even after exposure to Tat [69].
The role of astrocytes in HIV neuropathogenesis remains controversial. Some research suggests that astrocytes endocytose HIV-1 in order to sequester the virus and prevent its replication [70]. At the same time, research in an SIV model showed that infected astrocytes exhibit changes in morphology, namely, decreased arborization and dendritic length in both white and gray astrocytes. This suggests that infected astrocytes may create interruptions in glial-neuronal signaling, leading to brain atrophy and the symptoms associated with HAND [71]. A synthesis of these findings may indicate that astrocytes are not simply passively infected, but in fact attempt to rid the brain of HIV only to fall prey to HIV.
Given recent findings, it seems very probable that HIV both directly and indirectly affects normal neuronal activity through altering normal neuronal pathways and encouraging neuroinflammation. It may be that neuroinflammation amplifies the direct effects of HIV, by facilitating HIV replication or reseeding. Alternatively, it may be an important marker for neurotoxicity or it may offer a distinct form of neurotoxicity. Tat may be a contributing factor in both the pro-inflammatory and direct cytotoxic effects of CNS HIV infection. Additionally, astrocytes have gained importance in the conversation on neuronal damage in HIV. It is likely that a combination of direct and indirect effects of HIV cause the neurotoxicity and dysfunction underlying HAND. Whether these effects stem from a common pathway, or whether they are unlinked, presents an important question for considering treatment options.
Blipping and Asymptomatic CSF Escape on cART
Blipping, in which well-controlled viral load increases suddenly from undetectable levels, even during good adherence to cART, can be observed in otherwise stable patients with HIV. This phenomenon has been attributed to changes in viral reservoir stability and has been modeled using stochastic models [72]. These blips might be responsible for the ongoing inflammation and damage observed in chronic infection, such that the infected patient’s viremia is never truly stably undetectable. Similarly, low-level detectable HIV RNA in the CSF, termed “asymptomatic CSF escape” has been identified in 10 % of patients on long-term stable treatment with undetectable plasma viral load by standard assays [73]. This is distinct from “symptomatic CSF escape” which likely represents progressive underlying encephalitis and has clear clinical consequences. It is unclear whether “asymptomatic CSF escape” represents a stable persistence of HIV release from reservoirs or evidence of ongoing HIV replication, and its role in HIV neuropathogenesis in the setting of cART remains unclear. Longitudinal studies are needed to better assess the course of this low-level detectable HIV RNA and to determine whether this may be a harbinger of longer term progressive CNS disease.
Biomarkers of CNS Disease in Humans
While in vitro and animal model experiments have been incredibly valuable in better understanding the neuropathogenesis of HIV, clinical work remains critical to understanding how HIV affects the human brain. Since direct brain tissue cannot readily be sampled in living HIV-infected individuals, the development of biomarkers of the progression of HAND has become more clinically relevant. In recent years, molecular and neuroimaging markers have been recognized as important biomarkers in HAND.
In particular, the CSF has emerged as a potentially important proxy for measurements of pathological processes in the CNS. Numerous studies have supported the examination of CSF HIV RNA, soluble CSF markers of immune activation, and markers of neuronal injury in clinical and research investigations of HAND [56, 74]. A new, highly sensitive assay for NFL has recently been shown not only to be elevated in the neuroasymptomatic stages of HIV infection but also in fact to be persistently abnormal in the setting of suppressive cART, suggesting ongoing subtle neuronal injury [75•]. Similarly, measurement of HIV RNA by single copy assays reveals low-level viral persistence in some patients on cART [76]. The fact that HIV by SCA correlates with low-grade elevation in markers of abnormal macrophage activation suggests that this detected HIV may be mechanistically significant. Finally, various neuroimaging techniques have indicated important changes in the brains of patients progressing towards HAND. MRI, DTI, PET, and MRS have emerged as methods that might offer useful structural, metabolism, and metabolite information in both primary and chronic HIV infection [77]. In cART-naive patients with primary HIV infection, MRS shows increases in metabolites associated with inflammation, gliosis, and neuronal damage. These markers are attenuated after cART initiation [26•].
Given the need to detect and address stages of HIV neuropathogenesis prior to the development of HAND, novel biomarkers continue to be explored. For example, asymmetric dimethylarginine (ADMA), a biomarker of endothelial dysfunction, is increased in patients with asymptomatic HIV infection [78] and could posit association with disruption of the blood-brain barrier and HAND. While the role of ADMA remains unknown, this is one of a group of novel biomarkers which needs to be explored to optimize assessment of HIV and guide HIV care, particularly in patients at risk for HAND.
Comorbidities and Adjunctive Therapies
In postmortem studies of patients on cART, viral load was significantly decreased in the brain as compared to those who were not on cART [79]. While HAD has significantly decreased with cART, HAND still persists in a large proportion of patients with well-controlled HIV [6], and substantial evidence suggests that the CNS remains abnormal even in the context of apparently successful systemic viral suppression (see Peluso review). Many challenges remain in optimizing HIV treatment, and questions persist regarding cART strategies that may be neuroprotective or will most effectively ameliorate accrued neurologic disease. In particular, the calibration of treatment for optimal CNS penetrance presents an important practical question for patients and clinicians, which has been extensively reviewed elsewhere [61, 80].
The incomplete resolution of HAND in patients on HIV treatment suggests a need for adjunctive therapies that go beyond the effects of cART. Anti-inflammatory medications including aspirin have been explored in patients with HIV and have been preliminarily found to decrease platelet aggregation and improve vascular function in patients with HIV [81]. A recent study of a rat model of hepatic encephalopathy examined whether ibuprofen decreased neuroinflammation and microglial activation, finding improved cognitive and motor function in treated animals [82]. Though HAND is distinct in etiology and pathogenesis from hepatic encephalopathy, the common feature of neuroinflammation supports a similar potential role for anti-inflammatories in adjunctive approaches to HAND.
Finally, comorbidities ranging from aging to substance abuse have been implicated as potentiators of HAND. For example, the dopamine release associated with increased drug use may be associated with increased virus uptake in macrophages [83]. Future studies will be critical to ensure optimal care for patients with diverse clinical histories.
Conclusions
After over 30 years, HIV neuropathogenesis continues to present a dynamic challenge and important questions for clinical and scientific communities. Promising advances in clinical, animal model, and in vitro research on viral entry and primary CNS infection, CNS reservoirs, and neuronal toxicity remain critical in solidifying our understanding of how HIV affects the CNS. In the process of better understanding the neuropathogenesis of HIV, many potentially promising targets for therapy have been identified. Given research suggesting roles for both neuroinflammation and neurotoxicity in the course of infection, it seems that a more comprehensive treatment for HIV might take into account and address both of these effects.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Levy RM, Bredesen DE, Rosenblum ML. Neurological manifestations of the acquired immunodeficiency syndrome (AIDS): experience at UCSF and review of the literature. J Neurosurg. 1985;62(4):475–95.
Navia BA, Price RW. The acquired immunodeficiency syndrome dementia complex as the presenting or sole manifestation of human immunodeficiency virus infection. Arch Neurol. 1987;44(1):65–9.
Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5(1):69–81.
Robertson KR, Smurzynski M, Parsons TD, Wu K, Bosch RJ, Wu J, et al. The prevalence and incidence of neurocognitive impairment in the HAART era. AIDS. 2007;21(14):1915–21.
Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69(18):1789–99.
Heaton RK, Clifford DB, Franklin Jr DR, Woods SP, Ake C, Vaida F, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75(23):2087–96.
Cysique LA, Brew BJ. Prevalence of non-confounded HIV-associated neurocognitive impairment in the context of plasma HIV RNA suppression. J Neurovirol. 2011;17(2):176–83.
Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986;19(6):525–35.
Gyorkey F, Melnick JL, Gyorkey P. Human immunodeficiency virus in brain biopsies of patients with AIDS and progressive encephalopathy. J Infect Dis. 1987;155(5):870–6.
Enting RH, Prins JM, Jurriaans S, Brinkman K, Portegies P, Lange JM. Concentrations of human immunodeficiency virus type 1 (HIV-1) RNA in cerebrospinal fluid after antiretroviral treatment initiated during primary HIV-1 infection. Clin Infect Dis. 2001;32(7):1095–9.
Spudich S, Gisslen M, Hagberg L, Lee E, Liegler T, Brew B, et al. Central nervous system immune activation characterizes primary human immunodeficiency virus 1 infection even in participants with minimal cerebrospinal fluid viral burden. J Infect Dis. 2011;204(5):753–60.
Ho DD, Sarngadharan MG, Resnick L, Dimarzoveronese F, Rota TR, Hirsch MS. Primary human T-lymphotropic virus type III infection. Ann Intern Med. 1985;103(6 (Pt 1)):880–3.
Scarpini E, Sacilotto G, Lazzarin A, Geremia L, Doronzo R, Scarlato G. Acute ataxia coincident with seroconversion for anti-HIV. J Neurol. 1991;238(6):356–7.
Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, et al. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992;42(9):1736–9.
Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis. 2012;206(2):275–82.
Liu Y, Tang XP, McArthur JC, Scott J, Gartner S. Analysis of human immunodeficiency virus type 1 gp160 sequences from a patient with HIV dementia: evidence for monocyte trafficking into brain. J Neurovirol. 2000;6 Suppl 1:S70–81.
Spudich S, González-Scarano F. HIV-1-related central nervous system disease: current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb Perspect Med. 2012;2(6):a007120.
Andras IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res. 2003;74(2):255–65.
Xu R, Feng X, Xie X, Zhang J, Wu D, Xu L. HIV-1 Tat protein increases the permeability of brain endothelial cells by both inhibiting occludin expression and cleaving occludin via matrix metalloproteinase-9. Brain Res. 2012;1436:13–9.
Awan FM, Anjum S, Obaid A, Ali A, Paracha RZ, Janjua HA. In-silico analysis of claudin-5 reveals novel putative sites for post-translational modifications: insights into potential molecular determinants of blood-brain barrier breach during HIV-1 infiltration. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2014;27:355–65.
Woollard SM, Bhargavan B, Yu F, Kanmogne GD. Differential effects of Tat proteins derived from HIV-1 subtypes B and recombinant CRF02_AG on human brain microvascular endothelial cells: implications for blood-brain barrier dysfunction. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2014;34(6):1047–59.
Campbell J, Autissier P, MacLean AG, Burdo T, Westmoreland S, Gonzalez G, et al. VLA-4 treatment blocks virus traffic to the gut and brain early, and stabilizes CNS injury late in infection. 20th Conference for Retroviruses and Opportunistic Infections; Atlanta, GA 2013.
Lentz MR, Kim WK, Kim H, Soulas C, Lee V, Venna N, et al. Alterations in brain metabolism during the first year of HIV infection. J Neurovirol. 2011;17(3):220–9.
Sailasuta N, Ross W, Ananworanich J, Chalermchai T, DeGruttola V, Lerdlum S, et al. Change in brain magnetic resonance spectroscopy after treatment during acute HIV infection. PLoS One. 2012;7(11):e49272.
Suh J, Sinclair E, Peterson J, Lee E, Kyriakides TC, Li FY, et al. Progressive increase in central nervous system immune activation in untreated primary HIV-1 infection. J Neuroinflammation. 2014;11(1):199.
Young AC, Yiannoutsos CT, Hegde M, Lee E, Peterson J, Walter R, et al. Cerebral metabolite changes prior to and after antiretroviral therapy in primary HIV infection. Neurology. 2014;83(18):1592–600. This study uses longitudinal brain magnetic spectroscopy in individuals identified during primary HIV infection to demonstrate that neuroinflammation progressively increases during the early stages of infection prior to the initiation of antiretroviral therapy.
Schnell G, Price RW, Swanstrom R, Spudich S. Compartmentalization and clonal amplification of HIV-1 variants in the cerebrospinal fluid during primary infection. J Virol. 2010;84(5):2395–407.
Marcondes MC, Morsey B, Emanuel K, Lamberty BG, Flynn CT, Fox HS. CD8+ T cells maintain suppression of simian immunodeficiency virus in the central nervous system. J Inf Dis. 2015;211(1):40-4.
Schmitz JE, Simon MA, Kuroda MJ, Lifton MA, Ollert MW, Vogel CW, et al. A nonhuman primate model for the selective elimination of CD8+ lymphocytes using a mouse-human chimeric monoclonal antibody. Am J Pathol. 1999;154(6):1923–32.
Strickland SL, Rife BD, Lamers SL, Nolan DJ, Veras NM, Prosperi MC, et al. Spatiotemporal dynamics of simian immunodeficiency virus brain infection in CD8+ lymphocyte-depleted rhesus macaques with neuroAIDS. J Gen Virol. 2014;95(Pt 12):2784–95.
Wong JK, Ignacio CC, Torriani F, Havlir D, Fitch NJ, Richman DD. In vivo compartmentalization of human immunodeficiency virus: evidence from the examination of pol sequences from autopsy tissues. J Virol. 1997;71(3):2059–71.
Ritola K, Robertson K, Fiscus SA, Hall C, Swanstrom R. Increased human immunodeficiency virus type 1 (HIV-1) env compartmentalization in the presence of HIV-1-associated dementia. J Virol. 2005;79(16):10830–4.
Pillai SK, Pond SL, Liu Y, Good BM, Strain MC, Ellis RJ, et al. Genetic attributes of cerebrospinal fluid-derived HIV-1 env. Brain. 2006;129(Pt 7):1872–83.
Schnell G, Joseph S, Spudich S, Price RW, Swanstrom R. HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog. 2011;7(10):e1002286.
Dahl V, Gisslen M, Hagberg L, Peterson J, Shao W, Spudich S, et al. An example of genetically distinct HIV type 1 variants in cerebrospinal fluid and plasma during suppressive therapy. J Infect Dis. 2014;209(10):1618–22. This study employed single genome sequencing methods to sequence HIV RNA from paired cerebrospinal fluid and plasma samples of patients on cART, demonstrating that in the setting of suppressive treatment, unique HIV sequences could be identified in the CSF as compared to blood compartments.
Clements JE, Babas T, Mankowski JL, Suryanarayana K, Piatak Jr M, Tarwater PM, et al. The central nervous system as a reservoir for simian immunodeficiency virus (SIV): steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J Infect Dis. 2002;186(7):905–13.
Barber SA, Gama L, Dudaronek JM, Voelker T, Tarwater PM, Clements JE. Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus–macaque model. J Infect Dis. 2006;193(7):963–70.
Zink MC, Brice AK, Kelly KM, Queen SE, Gama L, Li M, et al. Simian immunodeficiency virus-infected macaques treated with highly active antiretroviral therapy have reduced central nervous system viral replication and inflammation but persistence of viral DNA. J Infect Dis. 2010;202(1):161–70.
Canestri A, Lescure FX, Jaureguiberry S, Moulignier A, Amiel C, Marcelin AG, et al. Discordance between cerebral spinal fluid and plasma HIV replication in patients with neurological symptoms who are receiving suppressive antiretroviral therapy. Clin Infect Dis. 2010;50(5):773–8. In this paper, the investigators describe a phenomenon of symptomatic CSF escape, wherein during cART, patients with marked neurologic symptoms have HIV RNA detected in the CSF either at higher levels than in the plasma, or in the absence of plasma HIV RNA detection, providing proof-of-concept evidence that the CNS may serve as an autonomous source of ongoing viral replication despite systemically active cART.
Peluso MJ, Ferretti F, Peterson J, Lee E, Fuchs D, Boschini A, et al. Cerebrospinal fluid HIV escape associated with progressive neurologic dysfunction in patients on antiretroviral therapy with well-controlled plasma viral load. AIDS. 2012.
An SF, Groves M, Gray F, Scaravilli F. Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J Neuropathol Exp Neurol. 1999;58(11):1156–62.
Bissel SJ, Wang G, Trichel AM, Murphey-Corb M, Wiley CA. Longitudinal analysis of monocyte/macrophage infection in simian immunodeficiency virus-infected, CD8+ T-cell-depleted macaques that develop lentiviral encephalitis. Am J Pathol. 2006;168(5):1553–69.
Thompson KA, Cherry CL, Bell JE, McLean CA. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol. 2011;179(4):1623–9. Extensive pathologic studies of patients who died of non-HIV related causes during neuroasymptomatic infection reveal HIV DNA in cells of the CNS as well as widespread microglial activation, confirming that HIV infection and immune activation is present prior to development of encephalitis in human HIV infection.
Shikuma CM, Nakamoto B, Shiramizu B, Liang CY, DeGruttola V, Bennett K, et al. Antiretroviral monocyte efficacy score linked to cognitive impairment in HIV. Antivir Ther. 2012;17(7):1233–42.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–5.
Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38(4):792–804.
Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–45.
Henrich TJ, Hanhauser E, Marty FM, Sirignano MN, Keating S, Lee TH, et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: report of 2 cases. Ann Intern Med. 2014;161(5):319–27.
Ellis RJ, Gamst AC, Capparelli E, Spector SA, Hsia K, Wolfson T, et al. Cerebrospinal fluid HIV RNA originates from both local CNS and systemic sources. Neurology. 2000;54(4):927–36.
Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol. 2009;66(2):253–8.
Zhuang K, Leda AR, Tsai L, Knight H, Harbison C, Gettie A, et al. Emergence of CD4 independence envelopes and astrocyte infection in R5 simian-human immunodeficiency virus model of encephalitis. J Virol. 2014;88(15):8407–20. The authors highlight the emergence of CD4-independent viral envelopes in a SHIVE model of HIV and link this to astrocyte infection in the model. This HIV consistent model of astrocyte infection may shed light on glial infection and neurodegeneration in HIV.
Strazza M, Pirrone V, Wigdahl B, Nonnemacher MR. Breaking down the barrier: the effects of HIV-1 on the blood-brain barrier. Brain Res. 2011;1399:96–115.
Peluso MJ, Meyerhoff DJ, Price RW, Peterson J, Lee E, Young AC, et al. Cerebrospinal fluid and neuroimaging biomarker abnormalities suggest early neurological injury in a subset of individuals during primary HIV infection. J Infect Dis. 2013;207(11):1703–12.
Lentz MR, Kim WK, Lee V, Bazner S, Halpern EF, Venna N, et al. Changes in MRS neuronal markers and T cell phenotypes observed during early HIV infection. Neurology. 2009;72(17):1465–72.
Kaul D, Ahlawat A, Gupta SD. HIV-1 genome-encoded hiv1-mir-H1 impairs cellular responses to infection. Mol Cell Biochem. 2009;323(1–2):143–8.
Hagberg L, Cinque P, Gisslen M, Brew BJ, Spudich S, Bestetti A, et al. Cerebrospinal fluid neopterin: an informative biomarker of central nervous system immune activation in HIV-1 infection. AIDS Res Ther. 2010;7:15.
Kamat A, Lyons JL, Misra V, Uno H, Morgello S, Singer EJ, et al. Monocyte activation markers in cerebrospinal fluid associated with impaired neurocognitive testing in advanced HIV infection. J Acquir Immune Defic Syndr. 2012;60(3):234–43.
Airoldi M, Bandera A, Trabattoni D, Tagliabue B, Arosio B, Soria A, et al. Neurocognitive impairment in HIV-infected naive patients with advanced disease: the role of virus and intrathecal immune activation. Clin Dev Immunol. 2012;2012:467154.
Gill AJ, Kovacsics CE, Cross SA, Vance PJ, Kolson LL, Jordan-Sciutto KL, et al. Heme oxygenase-1 deficiency accompanies neuropathogenesis of HIV-associated neurocognitive disorders. J Clin Invest. 2014;124(10):4459–72.
Yilmaz A, Price RW, Spudich S, Fuchs D, Hagberg L, Gisslen M. Persistent intrathecal immune activation in HIV-1-infected individuals on antiretroviral therapy. J Acquir Immune Defic Syndr. 2008;47(2):168–73.
Peluso MJ, Spudich S. Treatment of HIV in the CNS: effects of antiretroviral therapy and the promise of non-antiretroviral therapeutics. Curr HIV/AIDS Rep. 2014;11(3):353–62.
Romani B, Engelbrecht S, Glashoff RH. Functions of Tat: the versatile protein of human immunodeficiency virus type 1. J Gen Virol. 2010;91(Pt 1):1–12.
Sagnier S, Daussy CF, Borel S, Robert-Hebmann V, Faure M, Blanchet FP, et al. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J Virol. 2015;89(1):615-25.
Moran LM, Fitting S, Booze RM, Webb KM, Mactutus CF. Neonatal intrahippocampal HIV-1 protein Tat injection: neurobehavioral alterations in the absence of increased inflammatory cytokine activation. Int J Dev Neurosci Off J Int Soc Dev Neurosci. 2014;38C:195–203.
Meulendyke KA, Queen SE, Engle EL, Shirk EN, Liu J, Steiner JP, et al. Combination fluconazole/paroxetine treatment is neuroprotective despite ongoing neuroinflammation and viral replication in an SIV model of HIV neurological disease. J Neurovirol. 2014. This group administered fluconazole/paroxetine after the acute phase of infection in an SIV model and found that protection from neurodegeneration could be achieved despite continued neuroinflammation.
Tewari M, Monika, Varghese RK, Menon M, Seth P. Astrocytes mediate HIV-1 Tat-induced neuronal damage via ligand-gated ion channel P2X7R. J Neurochem. 2014. doi:10.1111/jnc.12953
Bethel-Brown C, Yao H, Hu G, Buch S. Platelet-derived growth factor (PDGF)-BB-mediated induction of monocyte chemoattractant protein 1 in human astrocytes: implications for HIV-associated neuroinflammation. J Neuroinflammation. 2012;9:262.
Eugenin EA, Berman JW. Cytochrome C dysregulation induced by HIV infection of astrocytes results in bystander apoptosis of uninfected astrocytes by an IP3 and calcium-dependent mechanism. J Neurochem. 2013;127(5):644–51. This study describes a cellular mechanism for bystander astrocyte apoptosis, neuroprotective effects of HIV on infected astrocytes, and the combination of these effects in the generation of a CNS HIV reservoir.
Shin AH, Kim HJ, Thayer SA. Subtype selective NMDA receptor antagonists induce recovery of synapses lost following exposure to HIV-1 Tat. Br J Pharmacol. 2012;166(3):1002–17.
Chauhan A, Tikoo A, Patel J, Abdullah AM. HIV-1 endocytosis in astrocytes: a kiss of death or survival of the fittest? Neurosci Res. 2014;88C:16–22.
Lee KM, Chiu KB, Renner NA, Sansing HA, Didier PJ, MacLean AG. Form follows function: astrocyte morphology and immune dysfunction in SIV neuroAIDS. J Neurovirol. 2014;20(5):474–84.
Wang S, Rong L. Stochastic population switch may explain the latent reservoir stability and intermittent viral blips in HIV patients on suppressive therapy. J Theor Biol. 2014;360:137–48.
Eden A, Fuchs D, Hagberg L, Nilsson S, Spudich S, Svennerholm B, et al. HIV-1 viral escape in cerebrospinal fluid of subjects on suppressive antiretroviral treatment. J Infect Dis. 2010;202(12):1819–25.
Gisslen M, Hagberg L, Rosengren L, Brew BJ, Cinque P, Spudich S, et al. Defining and evaluating HIV-related neurodegenerative disease and its treatment targets: a combinatorial approach to use of cerebrospinal fluid molecular biomarkers. J Neuroimmune Pharm. 2007;2(1):112–9.
Jessen Krut J, Mellberg T, Price RW, Hagberg L, Fuchs D, Rosengren L, et al. Biomarker evidence of axonal injury in neuroasymptomatic HIV-1 patients. PloS one. 2014;9(2):e88591. This paper describes elevated levels of cerebrospinal fluid (CSF) neurofilament light chain (NFL), a marker of active axonal injury, in neuroasymptomatic HIV-infected subjects on suppressive antiretroviral therapy, suggesting ongoing neural injury despite systemically successful treatment.
Dahl V, Peterson J, Fuchs D, Gisslen M, Palmer S, Price RW. Low levels of HIV-1 RNA detected in the cerebrospinal fluid after up to 10 years of suppressive therapy are associated with local immune activation. AIDS. 2014;28(15):2251–8.
Ances BM, Hammoud DA. Neuroimaging of HIV-associated neurocognitive disorders (HAND). Curr Opin HIV AIDS. 2014;9(6):545–51. doi:10.1097/COH.0000000000000112.
Hudson CL, Zemlin AE, Ipp H. The cardiovascular risk marker asymmetric dimethylarginine is elevated in asymptomatic, untreated HIV-1 infection and correlates with markers of immune activation and disease progression. Ann Clin Biochem. 2014;51(Pt 5):568–75.
Langford D, Marquie-Beck J, de Almeida S, Lazzaretto D, Letendre S, Grant I, et al. Relationship of antiretroviral treatment to postmortem brain tissue viral load in human immunodeficiency virus-infected patients. J Neurovirol. 2006;12(2):100–7.
Yilmaz A, Gisslen M. Treatment of HIV in the central nervous system. Semin Neurol. 2014;34(1):14–20.
O’Brien M, Montenont E, Hu L, Nardi MA, Valdes V, Merolla M, et al. Aspirin attenuates platelet activation and immune activation in HIV-1-infected subjects on antiretroviral therapy: a pilot study. J Acquir Immune Defic Syndr. 2013;63(3):280–8.
Rodrigo R, Cauli O, Gomez-Pinedo U, Agusti A, Hernandez-Rabaza V, Garcia-Verdugo JM, et al. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology. 2010;139(2):675–84.
Gaskill PJ, Yano HH, Kalpana GV, Javitch JA, Berman JW. Dopamine receptor activation increases HIV entry into primary human macrophages. PLoS One. 2014;9(9):e108232.
Compliance with Ethics Guidelines
Conflict of Interest
Zaina Zayyad and Serena Spudich declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on HIV Pathogenesis and Treatment
Rights and permissions
About this article

Cite this article
Zayyad, Z., Spudich, S. Neuropathogenesis of HIV: From Initial Neuroinvasion to HIV-Associated Neurocognitive Disorder (HAND). Curr HIV/AIDS Rep 12, 16–24 (2015). https://doi.org/10.1007/s11904-014-0255-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11904-014-0255-3
Keywords
- HIV
- AIDS
- HIV-associated neurocognitive disorder (HAND)
- Asymptomatic neurocognitive impairment (ANI)
- Mild neurocognitive disorder (MND)
- HIV-associated dementia (HAD)
- AIDS dementia complex
- Cerebrospinal fluid (CSF)
- Central nervous system (CNS)
- Combination antiretroviral therapy (cART)
- Neopterin
- Neurofilament light chain (NFL)
- Magnetic resonance spectroscopy (MRS)
- Neuroinflammation
- CSF escape
- Neurotoxicity