
Abstract
Increased life expectancy due to improved efficacy of cART has uncovered an increased risk of age-related morbidities in HIV+ individuals and catalyzed significant research into mechanisms driving these diseases. HIV infection increases the risk of non-communicable diseases common in the aged, including cardiovascular disease, neurocognitive decline, non-AIDS malignancies, osteoporosis, and frailty. These observations suggest that HIV accelerates immunological ageing, and there are many immunological similarities with the aged, including shortened telomeres, accumulation of senescent T cells and altered monocyte phenotype/function. However, the most critical similarity between HIV+ individuals and the elderly, which most likely underpins the heightened risk of non-communicable diseases, is chronic inflammation and associated immune activation. Here, we review the similarities between HIV+ individuals and the aged regarding the pathogenesis of inflammatory diseases, the current evidence for mechanisms driving these processes and discuss current and potential therapeutic strategies for addressing inflammatory co-morbidity in HIV+ infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic inflammation is a hallmark of ageing and is recognized as a central mechanism driving many age-related diseases. Indeed, levels of inflammatory markers including IL-6, TNF, and high sensitivity C-reactive protein (hsCRP) are independently associated with non-communicable diseases including cardiovascular disease (CVD), frailty and neurocognitive decline (see Table 1 for summary). HIV infection induces significant inflammation and this is incompletely restored by combination antiretroviral therapy (cART) [1]; elevated levels of inflammatory markers TNF, IL-6 and hsCRP as well as markers of innate immune activation including soluble (s) CD14, sCD163, and CXCL10 persist in combination antiretroviral therapy (cART)-treated individuals despite sustained viral suppression [2-5].
When comparing HIV infection and ageing, the question arises as to whether HIV is accelerating normal immunological ageing processes or whether both conditions result in chronic inflammation and increased co-morbidities due to parallel but mechanistically different pathogeneses. This question is critical to inform how best to prevent these conditions both in the setting of HIV infection and in HIV seronegative individuals with underlying low level chronic inflammation. Here we review the similarities in inflammatory disease pathogenesis between HIV+ individuals and the aged, and explore causative mechanisms to identify whether lessons we have learned from healthy ageing can guide prevention of non-communicable co-morbidities in the HIV+ population. We start by defining inflammation and immune activation, as these are commonly confused, then focus on two major age-related morbidities, cardiovascular, and bone disease.
The Relationship between Inflammation, Immune Activation and Immunosenescence
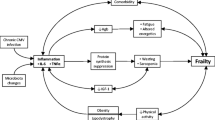
Whilst inflammation and immune activation are intimately related, these two parameters are discrete and are indicated by a distinct set of biomarkers. Inflammation is typically indicated by elevated plasma levels of pro-inflammatory cytokines (e.g., TNF, IL-6) and inflammatory markers such as the acute phase protein hsCRP. Soluble forms of the TNF receptors (TNFRI and TNFRII) are shed following TNF stimulation and are thus used as a biomarker of TNF-activation as levels are higher and more stable than TNF itself. Monocyte/macrophage activation biomarkers include the soluble lipopolysaccharide (LPS) receptor component CD14 (sCD14) which is shed following LPS stimulation [6], and neopterin and CXCL10, which are indicative of IFNγ-mediated activation [7, 8]. The function of the biomarker is not always consistent with how it is interpreted, such as with sCD163 which is used as a biomarker of inflammation-induced monocyte/macrophage activation, although CD163 itself exerts an anti-inflammatory effect [9]. Immune activation is typically measured by cellular markers such as HLA-DR and CD38 on T cells, CD40, CD80, and CD86 on dendritic cells [10] and CD11b on monocytes [11]. Markers of inflammation are often assumed to correlate with cellular activation, although this cannot always be assumed. Finally, chronic immune activation drives immunosenescence, which is indicated by differentiation/senescence markers such as CD57 and loss of CD28, and immune senescence can itself trigger further inflammation (discussed below) and immune dysfunction. The mechanistic links between inflammation, immune activation, and immune senescence (depicted in Fig. 1) likely vary under various pathological states and careful correlative analysis is required to define the most informative biomarkers for HIV-related non-communicable diseases.

Relationship between inflammation, immune activation and immune senescence
Cardiovascular Disease
HIV+ individuals have an increased risk of CVD, with a recent meta-analysis estimating the relative risk at 1.61 (95% CI 1.43–1.81) for untreated individuals and 2.00 (1.70–2.37) for those on antiretroviral therapy [12]. This risk is of similar magnitude to that attributed to the traditional CVD risk factors of male sex (HR 1.70; 1.32-2.18) and smoking (HR 2.35; 1.92-2.87) amongst HIV+ individuals in the DAD study [13]. Accurate determination of CVD risk in HIV+ individuals has been confounded by differences in treatment status, virologic suppression and differing adjustment for lifestyle and traditional risk factors. Whilst increased CVD risk persists after adjusting for traditional risk factors [14], the field still requires large-scale studies that adequately control for these and other risk factors known to influence CVD. Part of the increased CVD risk during HIV infection may be attributable to the effects of cART, as the relative risk in cART-treated individuals is 1.52 (95% CI 1.35-1.70) compared to untreated individuals [14]. Certain antiretroviral drugs including protease inhibitors (PIs) and abacavir have been implicated in increased CVD risk [15], however the link between abacavir and CVD remains controversial as associations seen in large cohort studies [16, 17] have not been confirmed in randomized control trials [18], and a recent meta-analyses found no association between abacavir and CVD [19]. Beyond this, there is an increased risk of CVD in HIV+ individuals which cannot be explained by treatment and traditional risk factors.
Chronic inflammation has an established role in the development of CVD in HIV+ and uninfected populations. In the general population, both IL-6 [20] and hsCRP [21] levels predict future cardiovascular events and increases in IL-6 levels in the elderly are associated with increased cardiovascular risk [22]. Interest in the role of inflammation in the development of CVD in HIV infection intensified following the results of the SMART study, which showed that interruptions to cART were associated with higher mortality from non-AIDS events than continuous therapy [23]. These findings were later linked to increased levels of the inflammatory markers IL-6, hsCRP and D-dimer [24]. Further analyses demonstrated that these markers were all independent predictors for future cardiovascular events, even when adjusted for known risk factors [25••].
The mechanism of inflammation-induced CVD involves endothelial activation. Arterial wall inflammation measured in vivo is heightened in HIV+ individuals with no known atherosclerosis compared to matched controls, but similar to HIV uninfected controls with known atherosclerosis, suggesting this may be an early stage in CVD development [26•]. Inflammatory cytokines act on endothelial cells to induce expression of adhesion receptors and increase chemokine production, which in turn promotes attachment and migration of leukocytes, particularly monocytes. A role for the chemokine receptor CCR5 in the development of atherosclerosis in HIV+ individuals is suggested by a link between carotid intima media thickness (cIMT, a clinical marker of atherosclerosis) and CCR5 mRNA levels in circulating leukocytes [27]. The antiretroviral drug maraviroc (a CCR5 antagonist) reduces inflammation-mediated recruitment of monocytes into plaques and inhibits plaque progression in a murine model [28], suggesting its therapeutic potential. However, pilot studies of cART intensification with maraviroc in poor immunological responders have yielded conflicting results regarding its effect on immune activation [29, 30], with one study showing maraviroc actually increased the proportion of HLA-DR+/CD38+ T cells [30]. Some of these discrepancies may be due to differences in immunophenotyping protocols, and further work with validated processing protocols is required to determine the effect of CCR5 antagonists on immune activation.
The migration of monocytes into developing atherosclerotic plaques and their development into inflammatory, lipid–laden foam cells is a critical early step in atherosclerosis (reviewed in [31]). Using an in vitro model of transendothelial migration, we have recently shown that monocytes from HIV+ individuals show an increased propensity to become foam cells and be retained in a model of sub-endothelial plaques (Maisa et al. submitted). This appears to involve both intrinsic alterations to monocytes and soluble factors, as our data shows that blocking TNF ligation ameliorates foam cell formation. These data suggest that in addition to their effects on endothelial activation, inflammatory factors including TNF may also act on monocytes to potentiate early atherogenic processes. Pro-inflammatory CD16+ monocytes, which expand in the blood of both HIV+ individuals [3, 32-34] and the elderly [3, 34-37], have been associated with increased risk of cardiovascular events [38] and peripheral vascular disease [39] in seronegative populations. Amongst viremic HIV+ individuals the phenotype of monocytes, including proportions of monocyte subsets, is similar to that of uninfected individuals with acute coronary syndrome [33] and levels of sCD163 are associated with non-calcified atherosclerotic plaques in both HIV+ men [40] and women [32], suggesting monocyte activation may contribute to CVD during HIV infection. Together, these data suggest a key role for inflammatory cytokines and monocytes activation in the pathogenesis of CVD in both HIV positive and negative individuals.
Reduced Bone Mineral Density and Osteoporosis
HIV+ individuals also have reduced bone mineral density (BMD) and an increased incidence of osteoporosis (odds ratios of6.4 and 3.7, respectively) [41]. This manifests clinically as a two-four fold increased prevalence of fracture [42] and an increased rate of multiple fractures [43]. Like all HIV co-morbidities, the cause of BMD loss in HIV+ individuals is multifactorial; an increased prevalence of factors known to affect BMD including low body mass index (BMI), smoking, diet and HCV co-infection has confounded quantitation of the HIV-specific effect. However, HIV seropositivity remains significantly associated with reduced BMD after adjustment for factors such as BMI [44]. Vitamin D deficiency is a well known risk factor for bone disease in the general population and although the prevalence of vitamin D insufficiency/deficiency in HIV+ populations is high (50-90%) [45]), it is similar to that of the general population.
Bone modeling is mediated by osteoclasts that resorb bone and osteoblasts that promote bone formation. Osteoclasts are activated via the receptor activator of NFκB ligand (RANKL) that binds to RANK expressed on the osteoclast surface, whilst the decoy receptor osteoprotegerin (OPG) antagonizes RANKL action and promotes bone formation. Osteoclasts/blasts and hematopoetic stem cells are derived from the same bone marrow progenitors and osteoblasts have a significant regulatory effect on function of immune cells (reviewed in [46]). Inflammation deregulates the delicate balance between osteoclast/blast activity; pro-inflammatory cytokines including TNF and IL-6 increase RANKL production, which stimulates osteoclast activity and bone resorption (reviewed in [47]). Further evidence of inflammation in the pathogenesis of osteoporosis is illustrated by hsCRP levels being independently associated with low BMD and fractures [48••, 49]. Immune activation also contributes to altered bone formation, as activated T and B cells produce significant amounts of RANKL. Additionally, LPS is known to stimulate osteoclast production [50]. Both chronic inflammation and immune activation are thus well-recognized mediators of bone loss and reduced BMD is a feature of chronic inflammatory conditions including rheumatoid arthritis [51], inflammatory bowel disease [52], diabetes [53], and chronic hepatitis infection [54]. In this sense, HIV is not unique in inducing inflammation-induced BMD loss, although it remains unclear whether the biochemical processes driving this are identical in chronic inflammatory diseases and in healthy ageing.
In addition to the effects of inflammation and immune activation, HIV-specific factors also induce BMD loss in HIV+ individuals. In vitro and animal studies indicate that viral proteins including gp120 and Vpr can stimulate osteoclast activity and BMD loss [55, 56]. ARV drugs also contribute to BMD loss. cART initiation is associated with a 2-6% loss in BMD within the first 2 years, irrespective of drug regimen. Whilst PIs and tenofovir [44, 57] are associated with increased BMD loss in cART-treated individuals, BMD loss during cART initiation is thought to be primarily due to disruption of the osteo-immunological balance associated with immune-reconstitution. The effect of ARV toxicity and ART initiation on BMD loss has confounded investigation of the relationship between inflammation/immune activation and bone loss during HIV infection and only limited data are available. However, T cell activation and HLA-DR + CD4+/CD8+ T cells are independently associated with low BMD in the setting of HIV infection [58]. Longitudinal analyses, with careful control for the use of tenofovir and PIs, are needed to clarify the association between inflammatory factors and BMD in HIV+ individuals.
Mechanisms Driving Chronic Inflammation and Inflammatory Diseases: Similarities between HIV+ Individuals and the Elderly
The mechanisms contributing to chronic inflammation in HIV+ individuals are multi-factorial and have been recently reviewed elsewhere [1]. Here we focus on three mechanisms which appear to contribute to inflammation and related diseases in both HIV+ individuals and the elderly.
Microbial Translocation
Damage to the gut epithelium during HIV infection is thought to result in increased translocation of microbial products from the gut into the blood stream. The latter contribute to both immune activation and chronic low level inflammation in HIV+ individuals ([2] and reviewed in [59]). Microbial products including LPS [2, 60, 61] and 16s rDNA [62] are elevated in the blood of both untreated and cART-treated HIV+ individuals. The soluble form of the TLR4 co-receptor CD14 (sCD14), is shed from the surface of monocytes upon activation by LPS [63] and we and others have reported that circulating sCD14 levels are also elevated in HIV+ individuals [32, 60, 61]. Levels of LPS, sCD14 and 16s rDNA levels correlate with traditional inflammatory markers including hsCRP [60, 64•, 65], IL-6 [60, 64•, 66], TNF [66], and D-dimer [60, 65], supporting the hypothesis that microbial translocation and resultant immune activation drive inflammation in the setting of HIV infection.
Following cART initiation, elevated LPS and sCD14 levels decrease [61] but do not normalize [67]. sCD14 and LPS levels reportedly correlate in many [61, 66, 68] but not all [3, 69] studies and recent work suggests that sCD14 and LPS levels may correlate only in patients with low CD4+ T cell counts and high HIV viral loads [65], suggesting these two markers should not be used indistinctly as measures of the same process.
Markers of microbial translocation are predictive of disease progression and mortality in HIV+ individuals [60, 70]. Amongst cART-treated individuals, elevated LPS levels are associated with reduced brachial artery flow mediated dilatation, indicating endothelial dysfunction [71], elevated cholesterol and decreased insulin sensitivity [72]. Prospectively, elevated LPS and sCD14 are independent predictors of future hypertension in cART naïve individuals [73] and of progression of subclinical atherosclerotic plaques amongst treated individuals, but not healthy controls [69]. Collectively, the available data suggest a role for microbial translocation in the development of CVD in HIV+ individuals.
There has been substantially less focus on microbial translocation during healthy ageing although recent evidence suggests its relevance. We and others have reported elevated plasma LPS [35] and sCD14 [67] levels amongst healthy, older individuals. A cross-sectional analysis of over 5000 individuals found an increase in sCD14 with age and demonstrated that baseline sCD14 predicted future cardiovascular events and mortality, independent of traditional cardiovascular risk factors [64•], suggesting a significant role of microbial translocation in the development of CVD in the elderly.
Interpretation of these findings is limited by the difficulty in measuring microbial translocation, particularly the unreliability of LPS assays. The Limulus Amebocyte Lysate (LAL) assay used to measure LPS and PCR amplification of16s rDNA are both highly susceptible to contamination by bacterial products, whilst LPS detection in serum and plasma is limited by the presence of inhibitors. There is substantial variation between individuals [74] and between serum versus plasma [75] regarding the extent to which inhibitors affect LPS detection. Heat inactivation and sample dilution can partially overcome these effects [74, 75]. However, a recent study using HIV+ samples suggested that dilutions of plasma as low as 1:500 (greater than the commonly used dilutions of 1:5–10) may be required to overcome LPS inhibition [74]. The detection of sCD14 is technically more reproducible than that of LPS, however, it is a marker of monocyte activation and thus an indirect measure of microbial translocation. There is also a substantial genetic contribution to sCD14 levels that accounts for 33% of variation [64•]. Thus, methodological differences and variations in assay performance could account for some of the discrepancies observed in the literature.
Telomeres
Telomeres are short, repetitive nucleotide sequences located at the ends of chromosomes, protecting them from degradation. Telomere shortening is a biological marker of ageing; shortened telomeres have been associated with age-related diseases including CVD diabetes and cancers (reviewed in [76] and [77]). A large prospective study of 19,838 subjects in Denmark found modest but significant associations between shortened telomeres and myocardial infarction, ischemic heart disease and early death [78]. Telomere length is an independent risk factor for CVD outcomes in the general population [79]. Telomere length correlates inversely with cIMT after adjustment for age [80] and also with markers of diabetes [81].
Ageing is characterized by an accumulation of late-differentiated T cells with both shortened telomeres and a senescent phenotype (see section below). This population is also expanded in HIV infection [82]. These senescent T cells have a heightened production of TNF, IL-6 and RANKL [83], potentially contributing to chronic inflammation and increased bone resorption in both HIV+ individuals and the aged. It is unclear whether telomere shortening and inflammation are mechanistically connected or whether both are indicative of an associated process, however the two phenomena occur concurrently. Telomere shorting in HIV+ individuals has been reported in T cells [84], and monocytes [35]. Given monocytes are not thought to undergo significant cell division in the periphery, shortened telomeres in peripheral blood monocytes is an unexpected finding and suggests shortening occurs within bone marrow precursor cells, a finding which may have implications for other cell types arising from these precursors including osteoclasts.
The mechanism underlying shortening of telomeres in HIV+ individuals may relate to diminished effects of telomerase (the enzyme responsible for maintaining telomere length) [84]; the normal up-regulation of telomerase in response to cell stimulation is also defective in HIV+ individuals [85]. Nucleot(s)idereverse transcriptase inhibitors (NRTIs) impair telomerase activity both in vitro and in vivo [84, 86]. NRTIs act as substrates for not only HIV reverse transcriptase but also telomerase and mitochondrial DNA polymerase γ (polγ). Our findings from a small cohort study showed that HIV+ patients receiving NRTIs had significantly shorter telomeres than individuals receiving non-NRTI-containing regimens or uninfected controls [86]. Inhibition of the telomerase reverse transcriptase (TERT) may have additional effects independent of telomere length, as the RT component of telomerase helps protect the mitochondria from oxidative stress (reviewed in [87]). The HIV proteins Tat [88] and Vpr [89] can inhibit telomerase in vitro although interestingly, Vpr mutants from long term non-progressors do not degrade TERT [89].
A mechanism of telomerase inhibition that may be common to HIV+ individuals and the elderly is inflammation, as TNF impairs telomerase activity in CD4+ [90] and CD8+ T cells [91] in vitro. We have also shown that monocytes, including the pro-inflammatory CD16+ monocyte subset, from both young HIV+ individuals and aged seronegative individuals show heightened basal and LPS-stimulated production of pro-inflammatory cytokines and both groups show shortened telomeres [3]. Thus, inflammation-induced telomere shortening and the production of pro-inflammatory cytokines by senescent T cells and activated monocytes may represent a positive feedback loop driving further immune senescence and inflammation in both HIV+ and aged populations.
Cytomegalovirus (CMV)
CMV infection plays a significant role in driving immune ageing and senescence and causes an expansion of late differentiated T cells in aged individuals. In the aged, up to 27% of total CD8+ T cells are specific for a small number of CMV epitopes [92]. Recent findings suggest the expansion of late differentiated CD28- memory T cells previously attributed to ageing is predominantly driven by CMV infection [93••, 94]. CMV infection induces pro-inflammatory cytokine release in vitro and serum CMV IgG levels correlate with inflammatory markers (in [95]). CMV-seropositivity has also been associated with increased risk of CVD [96] and mortality in CVD patients [97].
CMV-specific CD8+ T cells are expanded in HIV+ individuals to almost twice the level as uninfected controls, and this persists despite cART-treatment [98]. In HIV+ individuals, serum levels of CMV IgG are elevated and are associated with subclinical CVD [99, 100], whilst CMV-specific T cell responses are independently associated with cIMT [101]. A study of chronically infected HIV+ individuals in Thailand found 26% of treatment-naïve participants had detectable CMV DNA (a marker of CMV reactivation) [102]. Taken together, these data suggest HIV infection, either directly or indirectly, reactivates CMV and increases the immunological burden resulting from infection with this virus. It is possible that many of the age-related immunological effects of HIV may actually be secondary to, or at the very least confounded by, HIV-induced CMV-reactivation. However whilst direct causality to these diseases, including CVD, has not been demonstrated, inhibition of CMV with valganciclovir therapy in cART-treated HIV+ individuals with low CD4+ T cell counts mediated a significant reduction in CMV DNA and CD8+ T cell activation [103].
Delineating the contribution of CMV to inflammation and immune activation during both HIV infection and ageing is difficult as CMV seropositivity is ubiquitous in both groups (70-80% CMV seropositivity in HIV-negative individuals aged >40 years and >90% in HIV+ individuals) and is rarely controlled for in cohort studies. Serology for CMV has limited value in providing evidence of reactivation. However quantification of CMV reactivation remains challenging as CMV DNA viremia is rarely detected in healthy individuals, is expensive to monitor, and CMV-specific T cell responses do not always correlate with viremia. Whilst CMV-specific T cell responses are more reflective of viral burden and more likely to indicate reactivation than seropositivity a more sensitive and reliable test for CMV reactivation is needed to help clarify the above-mentioned issues.
T-cell Activation and Comorbidities in cART Treated Patients
Immune senescence involves changes to many immune cell types, but is often measured as the accumulation of highly differentiated T-cells, particularly in the CD8+ T-cell compartment (reviewed in [104]. T-cell senescence is characterized by the loss of the co-stimulatory molecules CD27 and CD28, expression of CD57, impaired proliferation, shorter telomeres, as well as the secretion of pro-inflammatory cytokines IL-1, IL-6, and TNF (reviewed in [105, 106]. CD8+ T-cell activation (CD38 + HLADR+) and senescence (CD57 + CD28-) have been associated with markers of atherosclerosis and vascular dysfunction in cART treated patients [107-109] although more recent studies which adjust for other markers have found markers of innate immune activation to be more important [110-112]. There is also a reported association in cART-treated HIV+ patients between CD8+ T-cell senescence (but not T cell activation) and Kaposi’s sarcoma [113]. Although the role of T-cell activation in the development of comorbidities in HIV infection is currently unclear, numerous studies have consistently shown that deficiencies in the number of circulating CD4+ T-cells post-cART is a strong risk factor for multiple age-related co-morbidities including CVD, osteoporosis, non-AIDS related malignancies, and frailty in cART treated individuals [114-118]. These data collectively imply that the adaptive immune system may have a more indirect role in driving clinical end-points in HIV+ individuals.
Ageing with HIV in Resource Limited Settings
The majority of studies investigating age-related comorbidities in HIV+ population have been from developed countries [119-121] and there are limited data from resource-limited settings. However, the prevalence of non-communicable diseases worldwide is high, and is responsible for substantial mortality in both resource rich and poor countries [122-127]. In China, non-communicable diseases account for 80% of all deaths and 70% of total disability-adjusted life-years lost [128]. In North-eastern China, 29% of the urban population has hypertension with adequate control only in 4% [125].
A similarly high prevalence of comorbidities has also been reported among HIV+ individuals living in developing countries. Approximately 70% of over 5000 women recruited for a recent HIV prevention trial in Kwazulu Natal, Africa (median age 27 years) were either overweight or obese [123], with a similar proportion of HIV+ women in South Africa also obese [129] and the prevalence of hypertension among HIV+ individuals in some African countries ranging from 10-45% [130-132]. In HIV+ individuals (median age 37 years) receiving cART in Taiwan, the combined prevalence of osteoporosis/osteopenia was 40% [133]. These data collectively suggest that despite differences in risk exposures between developed and developing country settings, non-communicable diseases are a growing concern and are already presenting a major disease burden both in the HIV+ and uninfected populations in resource limited settings.
Many of the HIV-related risk factors known to be associated with premature aging in HIV including advanced immunodeficiency [116, 117], chronic immune activation and inflammation [134] and chronic co-infections [135, 136] are all prevalent in the resource limited setting [137-141]. Most HIV+ people residing in these countries are from poor socioeconomic backgrounds, an independent factor associated with accelerated aging [142]. Whilst there is improved access to ART, and globally HIV+ patients are living longer, there is generally poor integration of health services in most HIV health care facilities in these settings [143, 144]. Thus patients are likely to present with non-communicable diseases only after the development of significant morbidity. A greater understanding of the extent and risk factors for age-related comorbidities in the HIV+ population in resource limited settings as well as the magnitude of specific pathogenetic factors that may contribute to chronic inflammation and immune activation (e.g., chronic parasitemia) is urgently needed in order to assist public health efforts to integrate non-communicable disease management into existing HIV prevention and treatment programs.
Therapeutics
Preventing inflammatory disease by addressing disease risk factors such as smoking, hypertension, obesity, vitamin D deficiency, are equally important in both the HIV+ and general populations, but given the significant evidence of shared disease pathogenesis, the question arises as to whether different therapeutic approaches are required to specifically target these non-communicable morbidities in HIV+ individuals, or whether we can learn lessons from healthy ageing.
Preventing inflammatory diseases by reducing inflammation seems an obvious approach, although anti-inflammatory drugs are used cautiously in the aged due to adverse effects including gastrointestinal bleeding. Low-dose aspirin is widely used for CVD prophylaxis in the general population; its ability to prevent other age-related inflammatory diseases is unproven. The efficacy of low dose (100 mg daily) aspirin treatment for 5 years in preventing a range of age-related conditions is currently being evaluated in a cohort of 19,000 individuals aged >70 years in the Aspirin in reducing events in the elderly (ASPREE) trial [145]. Results from this study may have relevance also in the HIV+ population. There are promising results from a 1 week trial of low-dose aspirin (325 mg loading dose then 81 mg daily) in 25 cART-treated HIV+ individuals [146•] where a significant reduction in T cell activation and plasma levels of sCD14 but not hsCRP, D-dimer and IL-6, were noted. These anti-inflammatory effects were hypothesized to be secondary to reduced platelet activation and thus monocyte activation, as the aspirin dose used was well below that required to mediate anti-inflammatory effects (3–4 mg daily).
Whilst statins are widely used in the general population as cholesterol-lowering drugs, they also have immunologic and cardioprotective effects including improved endothelial function and reduced T cell activation as well as having anti-inflammatory effects (for review see [147]). Trials evaluating the anti-inflammatory efficacy of statins in HIV+ individuals show mixed results. Rosuvastatin, atorvastatin, and pravastatin can significantly reduce serum IL-6, TNF and hsCRP in cART-treated HIV+ individuals [148], however anti-inflammatory effects of pravastatin were not supported in a further trial [149]. Other studies suggest statins can reduce immune activation without altering markers of inflammation [150, 151]. Statin therapy is associated with reduced all-cause mortality in HIV+ individuals with pre-diagnosed co-morbidity, but the benefit in those without co-morbidity seems less significant [152]. Larger randomized trials in HIV+ individuals are required to clarify their beneficial effect in ameliorating inflammation and inflammatory diseases.
Although chronic endotoxemia is present in both HIV+ individuals and the aged, elevated plasma LPS levels in young HIV+ individuals [3], suggesting that agents that inhibit LPS signaling may be of benefit in these patients. A number of trials have investigated the efficacy of TLR inhibitors chloroquine (CQ) and hydroxychloroquine (HCQ) to reduce immune activation and resultant inflammation. These agents block endosomal acidification and thus activation of the endosomal TLR receptors TLR3, 7, and 9. In a small study, CQ/HCQ reduced CD4+, CD8+ T cell, and CD14+ monocyte activation and plasma LPS and IL-6 levels in cART-treated non-immunologic responders [153, 154]. In contrast, HCQ use in treatment-naïve participants with CD4+ T cell counts >400 cells/ml did not significantly reduce CD8+ T cell activation or inflammation; in fact CD4+ T cell decline and plasma viral load were significantly increased in the HCQ arm [155]. These results suggest that inhibition of TLR responses in viremic HIV+ individuals may have adverse effects on HIV control and suggest these drugs might only be cautiously used in individuals with virologic suppression.
In addition to increased microbial translocation, HIV+ individuals have an altered gut microbiota, with increased concentrations of P. aeruginosa and C. Albicans and reduced concentrations of Bifidobacteria and Lactobacilli (reviewed in [156]). The gut microbiota has significant influence on mucosal immunity, and treatment of SIV-infected macaques with prebiotics in addition to ART improves gastrointestinal immunity [157], although limited data from HIV+ individuals show inconsistent effects on markers of inflammation and immune activation [158, 159]. As rifamycins have an anti-inflammatory effect and can inhibit LPS-induced cytokine production trials are currently underway to determine the efficacy of non-absorbed antibiotics such as rifaximin in reducing microbial translocation during HIV infection. Given the central role that microbial translocation is purported to play in HIV-related inflammation/immune activation, significantly more data on the efficacy of pro/prebiotics and related therapeutics in both viremic and virologically suppressed HIV+ individuals are warranted.
Conclusion
The pathogenesis of inflammation-driven, non-communicable diseases in HIV+ individuals is complex and multi-factorial, and whilst there are clear HIV-specific mechanisms contributing to certain co-morbidities, it is equally clear that chronic inflammation drives many of these diseases in HIV+ diseases as well as in healthy ageing. Although the etiologies vary, mechanisms contributing to inflammation such as microbial translocation, immune activation and dysfunction and immune senescence act in parallel in both HIV+ individuals and the aged. Elucidating which manifestations of HIV co-morbidities require novel interventions, and which will benefit from traditional prevention and treatment strategies, is a priority area for research as the HIV+ population ages in both western society and in resource limited settings.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Deeks SG, Tracy R, Douek DC. Systemic Effects of Inflammation on Health during Chronic HIV Infection. Immunity. 2013;39:633–45.
Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71.
Hearps AC, Maisa A, Cheng WJ, et al. HIV infection induces age-related changes to monocytes and innate immune activation in young men that persist despite combination antiretroviral therapy. AIDS. 2012;26:843–53.
French MA, King MS, Tschampa JM, da Silva BA, Landay AL. Serum immune activation markers are persistently increased in patients with HIV infection after 6 years of antiretroviral therapy despite suppression of viral replication and reconstitution of CD4+ T cells. J Infect Dis. 2009;200:1212–5.
Burdo TH, Lentz MR, Autissier P, et al. Soluble CD163 made by monocyte/macrophages is a novel marker of HIV activity in early and chronic infection prior to and after anti-retroviral therapy. J Infect Dis. 2011;204:154–63.
Bazil V, Strominger JL. Shedding as a mechanism of down-modulation of CD14 on stimulated human monocytes. J Immunol. 1991;147:1567–74.
Murr C, Widner B, Wirleitner B, Fuchs D. Neopterin as a marker for immune system activation. Curr Drug Metab. 2002;3:175–87.
Neville LF, Mathiak G, Bagasra O. The immunobiology of interferon-gamma inducible protein 10 kD (IP-10): a novel, pleiotropic member of the C-X-C chemokine superfamily. Cytokine Growth Factor Rev. 1997;8:207–19.
Kowal K, Silver R, Slawinska E, et al. CD163 and its role in inflammation. Folia Histochem Cytobiol. 2011;49:365–74.
Hart DN. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood. 1997;90:3245–87.
Lundahl J, Hallden G, Skold CM. Human blood monocytes, but not alveolar macrophages, reveal increased CD11b/CD18 expression and adhesion properties upon receptor-dependent activation. Eur Respir J. 1996;9:1188–94.
Islam FM, Wu J, Jansson J, Wilson DP. Relative risk of cardiovascular disease among people living with HIV: a systematic review and meta-analysis. HIV Med. 2012;13:453–68.
Friis-Moller N, Thiebaut R, Reiss P, et al. Predicting the risk of cardiovascular disease in HIV-infected patients: the data collection on adverse effects of anti-HIV drugs study. Eur J Cardiovasc Prev Rehabil. 2010;17:491–501.
Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab. 2007;92:2506–12.
Sabin CA, Worm SW, Weber R, et al. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients enrolled in the D:A:D study: a multi-cohort collaboration. Lancet. 2008;371:1417–26.
group SIsgaDADs. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients. AIDS. 2008;22:F17–24.
Worm SW, Sabin C, Weber R, et al. Risk of myocardial infarction in patients with HIV infection exposed to specific individual antiretroviral drugs from the 3 major drug classes: the data collection on adverse events of anti-HIV drugs (D:A:D) study. J Infect Dis. 2010;201:318–30.
Ribaudo HJ, Benson CA, Zheng Y, et al. No risk of myocardial infarction associated with initial antiretroviral treatment containing abacavir: short and long-term results from ACTG A5001/ALLRT. Clin Infect Dis. 2011;52:929–40.
Ding X, Andraca-Carrera E, Cooper C, et al. No association of abacavir use with myocardial infarction: findings of an FDA meta-analysis. J Acquir Immune Defic Syndr. 2012;61:441–7.
Danesh J, Kaptoge S, Mann AG, et al. Long-term interleukin-6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PLoS Med. 2008;5:e78.
Ridker PM. High-Sensitivity C-Reactive Protein: Potential Adjunct for Global Risk Assessment in the Primary Prevention of Cardiovascular Disease. Circulation. 2001;103:1813–8.
Jenny NS, French B, Arnold AM, et al. Long-term assessment of inflammation and healthy aging in late life: the Cardiovascular Health Study All Stars. J Gerontol A Biol Sci Med Sci. 2012;67:970–6.
El-Sadr WM, Lundgren J, Neaton JD, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355:2283–96.
Kuller LH, Tracy R, Belloso W, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5:e203.
Duprez DA, Neuhaus J, Kuller LH, et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PLoS One. 2012;7:e44454. This large-scale analysis of cardiovascular events in the SMART study demonstrated that the inflammatory markers IL-6, D-dimer and hsCRP confer an increased risk of cardiovascular events amongst treated, HIV infected individuals, independent of other risk factors. This important finding provides evidence for the role of inflammation in the development of non-AIDS comorbidities in HIV infected individuals.
Subramanian S, Tawakol A, Burdo TH, et al. Arterial inflammation in patients with HIV. JAMA. 2012;308:379–86. This study demonstrates that HIV infected individuals have higher levels of arterial wall inflammation compared to controls matched for other cardiovascular risk factors, implicating inflammation as a plausible mechanism for the increased cardiovascular risk conferred by HIV infection.
Fernandez-Sender L, Alonso-Villaverde C, Rull A, et al. A possible role for CCR5 in the progression of atherosclerosis in HIV-infected patients: a cross-sectional study. AIDS Res Ther. 2013;10:11.
Cipriani S, Francisci D, Mencarelli A, et al. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation. 2013;127:2114–24.
Wilkin TJ, Lalama CM, McKinnon J, et al. A pilot trial of adding maraviroc to suppressive antiretroviral therapy for suboptimal CD4(+) T-cell recovery despite sustained virologic suppression: ACTG A5256. J Infect Dis. 2012;206:534–42.
Hunt PW, Shulman NS, Hayes TL, et al. The immunologic effects of maraviroc intensification in treated HIV-infected individuals with incomplete CD4+ T-cell recovery: a randomized trial. Blood. 2013;121:4635–46.
Crowe SM, Westhorpe CL, Mukhamedova N, et al. The macrophage: the intersection between HIV infection and atherosclerosis. J Leukoc Biol. 2010;87:589–98.
Fitch KV, Srinivasa S, Abbara S, et al. Noncalcified Coronary Atherosclerotic Plaque and Immune Activation in HIV-infected Women. J Infect Dis. 2013;208:1737–46.
Funderburg NT, Zidar DA, Shive C, et al. Shared monocyte subset phenotypes in HIV-1 infection and in uninfected subjects with acute coronary syndrome. Blood. 2012;120:4599–608.
Martin GE, Gouillou M, Hearps AC, et al. Age-associated changes in monocyte and innate immune activation markers occur more rapidly in HIV infected women. PLoS One. 2013;8:e55279.
Hearps AC, Martin GE, Angelovich TA, et al. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell. 2012;11:867–75.
Nyugen J, Agrawal S, Gollapudi S, Gupta S. Impaired functions of peripheral blood monocyte subpopulations in aged humans. J Clin Immunol. 2010;30:806–13.
Seidler S, Zimmermann HW, Bartneck M, Trautwein C, Tacke F. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 2010;11:30.
Rogacev KS, Cremers B, Zawada AM, et al. CD14++CD16+ Monocytes Independently Predict Cardiovascular Events: A Cohort Study of 951 Patients Referred for Elective Coronary Angiography. J Am Coll Cardiol. 2012;60:1512–20.
Dopheide JF, Obst V, Doppler C, et al. Phenotypic characterisation of pro-inflammatory monocytes and dendritic cells in peripheral arterial disease. Thromb Haemost. 2012;108:1198–207.
Burdo TH, Lo J, Abbara S, et al. Soluble CD163, a Novel Marker of Activated Macrophages, Is Elevated and Associated With Noncalcified Coronary Plaque in HIV-Infected Patients. J Infect Dis. 2011;204:1227–36.
Brown TT, Qaqish RB. Antiretroviral therapy and the prevalence of osteopenia and osteoporosis: a meta-analytic review. Aids. 2006;20:2165–74.
Triant VA, Brown TT, Lee H, Grinspoon SK. Fracture prevalence among human immunodeficiency virus (HIV)-infected versus non-HIV-infected patients in a large U.S. healthcare system. J Clin Endocrinol Metab. 2008;93:3499–504.
Torti C, Mazziotti G, Soldini PA, et al. High prevalence of radiological vertebral fractures in HIV-infected males. Endocrine. 2012;41:512–7.
Yin MT, Zhang CA, McMahon DJ, et al. Higher rates of bone loss in postmenopausal HIV-infected women: a longitudinal study. J Clin Endocrinol Metab. 2012;97:554–62.
Pinzone MR, Di Rosa M, Malaguarnera M, et al. Vitamin D deficiency in HIV infection: an underestimated and undertreated epidemic. Eur Rev Med Pharmacol Sci. 2013;17:1218–32.
Ofotokun I, McIntosh E, Weitzmann MN. HIV: inflammation and bone. Curr HIV/AIDS Rep. 2012;9:16–25.
Zupan J, Jeras M, Marc J. Osteoimmunology and the influence of pro-inflammatory cytokines on osteoclasts. Biochem Med (Zagreb). 2013;23:43–63.
de Pablo P, Cooper MS, Buckley CD. Association between bone mineral density and C-reactive protein in a large population-based sample. Arthritis Rheum. 2012;64:2624–31. A large study investigating the realtionship between hs-CRP levels and BMD in a cohort of 10,475 individuals. hsCRP levels were significantly and inversely associated with BMD after adjustment for a range of demographic and lifestyle factors including age, poverty, menopause, body mass index, smoking, co-morbidities etc. The large particiant numbers and extensive adjustment for confounders provide strong evidence for a link between inflammation and reduecd BMD.
Eriksson AL, Moverare-Skrtic S, Ljunggren O, et al.: High sensitive CRP is an independent risk factor for all fractures and vertebral fractures in elderly men: The MrOS Sweden study. J Bone Miner Res. 2013. doi:10.1002/jbmr.2037.
Zou W, Bar-Shavit Z. Dual modulation of osteoclast differentiation by lipopolysaccharide. J Bone Miner Res. 2002;17:1211–8.
Matuszewska A, Szechinski J. Evaluation of selected bone metabolism markers in rheumatoid arthritis patients. Adv Clin Exp Med. 2013;22:193–202.
Ghishan FK, Kiela PR. Advances in the understanding of mineral and bone metabolism in inflammatory bowel diseases. Am J Physiol Gastrointest Liver Physiol. 2011;300:G191–201.
Merlotti D, Gennari L, Dotta F, Lauro D, Nuti R. Mechanisms of impaired bone strength in type 1 and 2 diabetes. Nutr Metab Cardiovasc Dis. 2010;20:683–90.
Lin JC, Hsieh TY, Wu CC, et al. Association between chronic hepatitis C virus infection and bone mineral density. Calcif Tissue Int. 2012;91:423–9.
Vikulina T, Fan X, Yamaguchi M, et al. Alterations in the immuno-skeletal interface drive bone destruction in HIV-1 transgenic rats. Proc Natl Acad Sci U S A. 2010;107:13848–53.
Walker Harris V, Brown TT. Bone loss in the HIV-infected patient: evidence, clinical implications, and treatment strategies. J Infect Dis. 2012;205 Suppl 3:S391–8.
Hoy J. Bone, fracture and frailty. Curr Opin HIV AIDS. 2011;6:309–14.
Gazzola L, Bellistri GM, Tincati C, et al. Association between peripheral T-Lymphocyte activation and impaired bone mineral density in HIV-infected patients. J Transl Med. 2013;11:51.
Marchetti G, Tincati C, Silvestri G. Microbial Translocation in the Pathogenesis of HIV Infection and AIDS. Clin Microbiol Rev. 2013;26:2–18.
Sandler NG, Wand H, Roque A, et al. Plasma Levels of Soluble CD14 Independently Predict Mortality in HIV Infection. J Infect Dis. 2011;203:780–90.
Rajasuriar R, Booth D, Solomon A, et al. Biological determinants of immune reconstitution in HIV-infected patients receiving antiretroviral therapy: the role of interleukin 7 and interleukin 7 receptor alpha and microbial translocation. J Infect Dis. 2010;202:1254–64.
Jiang W, Lederman MM, Hunt P, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199:1177–85.
Kitchens RL, Thompson PA. Modulatory effects of sCD14 and LBP on LPS-host cell interactions. J Endotoxin Res. 2005;11:225–9.
Reiner AP, Lange EM, Jenny NS, et al. Soluble CD14: Genomewide Association Analysis and Relationship to Cardiovascular Risk and Mortality in Older Adults. Arterioscler Thromb Vasc Biol. 2013;33:158–64. This study provides strong evidence for sCD14 as an independent predictor of cardiovascular events in previously healthy, aged individuals. The long-term follow up and large study size make this the strongest evidence linking sCD14 to cardiovascular disease in any population.
Romero-Sanchez M, Gonzalez-Serna A, Pacheco YM, et al. Different biological significance of sCD14 and LPS in HIV-infection: importance of the immunovirology stage and association with HIV-disease progression markers. J Infect. 2012;65:431–8.
Reus S, Portilla J, Sanchez-Paya J, et al. Low-level HIV viremia is associated with microbial translocation and inflammation. J Acquir Immune Defic Syndr. 2013;62:129–34.
Méndez-Lagares G, Romero-Sánchez MC, Ruiz-Mateos E, et al. Long-term suppressive combined antiretroviral treatment does not normalize serum sCD14 levels. J Infect Dis. 2013;207(8):1221–5.
Ancuta P, Kamat A, Kunstman KJ, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One. 2008;3:e2516.
Kelesidis T, Kendall MA, Yang OO, Hodis HN, Currier JS. Biomarkers of microbial translocation and macrophage activation: association with progression of subclinical atherosclerosis in HIV-1 infection. J Infect Dis. 2012;206:1558–67.
Marchetti G, Cozzi-Lepri A, Merlini E, et al. Microbial translocation predicts disease progression of HIV-infected antiretroviral-naive patients with high CD4+ cell count. Aids. 2011;25:1385–94.
Blodget E, Shen C, Aldrovandi G, et al. Relationship between microbial translocation and endothelial function in HIV infected patients. PLoS ONE. 2012;7:e42624. Electronic Resource.
Pedersen KK, Pedersen M, Troseid M, et al. Microbial Translocation in HIV Infection is Associated with Dyslipidemia, Insulin Resistance, and Risk of Myocardial Infarction. J Acquir Immune Defic Syndr. 2013;64:425–33.
Manner IW, Baekken M, Kvale D, et al. Markers of microbial translocation predict hypertension in HIV-infected individuals. HIV Med. 2013;14:354–61.
Balagopal A, Gama L, Franco V, et al. Detection of microbial translocation in HIV and SIV infection using the Limulus amebocyte lysate assay is masked by serum and plasma. PLoS ONE. 2012;7:e41258. Electronic Resource.
Hurley JC. Endotoxemia: methods of detection and clinical correlates. Clin Microbiol Rev. 1995;8:268–92.
Nilsson PM, Tufvesson H, Leosdottir M, and Melander O: Telomeres and cardiovascular disease risk: an update 2013. Transl Res. 2013;162:371–80.
Kong CM, Lee XW, Wang X. Telomere shortening in human diseases. FEBS J. 2013;280:3180–93.
Weischer M, Bojesen SE, Cawthon RM, et al. Short telomere length, myocardial infarction, ischemic heart disease, and early death. Arterioscler Thromb Vasc Biol. 2012;32:822–9.
Willeit P, Willeit J, Brandstatter A, et al. Cellular aging reflected by leukocyte telomere length predicts advanced atherosclerosis and cardiovascular disease risk. Arterioscler Thromb Vasc Biol. 2010;30:1649–56.
Sanders JL, Fitzpatrick AL, Boudreau RM, et al. Leukocyte telomere length is associated with noninvasively measured age-related disease: The Cardiovascular Health Study. J Gerontol A Biol Sci Med Sci. 2012;67:409–16.
Fitzpatrick AL, Kronmal RA, Gardner JP, et al. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am J Epidemiol. 2007;165:14–21.
Effros RB, Allsopp R, Chiu CP, et al.: Shortened telomeres in the expanded CD28-CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS. 1996;10:F17–22.
Effros RB, Dagarag M, Spaulding C, Man J. The role of CD8+ T-cell replicative senescence in human aging. Immunol Rev. 2005;205:147–57.
Ballon G, Ometto L, Righetti E, et al. Human immunodeficiency virus type 1 modulates telomerase activity in peripheral blood lymphocytes. J Infect Dis. 2001;183:417–24.
Reynoso R, Laufer N, Bolcic F, Quarleri J. Telomerase activity in peripheral blood mononuclear cells from HIV and HIV-HCV coinfected patients. Virus Res. 2010;147:284–7.
Leeansyah E, Cameron PU, Solomon A, et al. Inhibition of telomerase activity by human immunodeficiency virus (HIV) nucleos(t)ide reverse transcriptase inhibitors: a potential factor contributing to HIV-associated accelerated aging. J Infect Dis. 2013;207:1157–65.
Bollmann FM. Telomerase inhibition may contribute to accelerated mitochondrial aging induced by anti-retroviral HIV treatment. Med Hypotheses. 2013;81:285–7.
Comandini A, Naro C, Adamo R, et al. Molecular mechanisms involved in HIV-1-Tat mediated inhibition of telomerase activity in human CD4(+) T lymphocytes. Mol Immunol. 2013;54:181–92.
Wang X, Singh S, Jung HY, et al. HIV-1 Vpr protein inhibits telomerase activity via the EDD-DDB1-VPRBP E3 ligase complex. J Biol Chem. 2013;288:15474–80.
Di Mitri D, Azevedo RI, Henson SM, et al. Reversible senescence in human CD4 + CD45RA + CD27- memory T cells. J Immunol. 2011;187:2093–100.
Parish ST, Wu JE, Effros RB. Modulation of T lymphocyte replicative senescence via TNF-{alpha} inhibition: role of caspase-3. J Immunol. 2009;182:4237–43.
Ouyang Q, Wagner WM, Wikby A, et al. Large numbers of dysfunctional CD8+ T lymphocytes bearing receptors for a single dominant CMV epitope in the very old. J Clin Immunol. 2003;23:247–57.
Derhovanessian E, Maier AB, Hahnel K, et al. Infection with cytomegalovirus but not herpes simplex virus induces the accumulation of late-differentiated CD4+ and CD8+ T-cells in humans. J Gen Virol. 2011;92:2746–56. This analysis of the effect of CMV and herpes simplex virus on T cell subsets in the aged demostrated that the expansion of CD28- memory T cells and ‘senescent’ CD57+ CD8+ T cells previously thought to be due to ageing is only seen in CMV seropositive individuals. This highlights the important roel CMV plays in immunosenescence and the need to adjust for CMV status, and ideally CMV reactivation, in studies of immune activation and ageing.
Chidrawar S, Khan N, Wei W, et al. Cytomegalovirus-seropositivity has a profound influence on the magnitude of major lymphoid subsets within healthy individuals. Clin Exp Immunol. 2009;155:423–32.
Solana R, Tarazona R, Aiello AE, et al. CMV and Immunosenescence: from basics to clinics. Immun Ageing. 2012;9:23.
Simanek AM, Dowd JB, Pawelec G, et al. Seropositivity to cytomegalovirus, inflammation, all-cause and cardiovascular disease-related mortality in the United States. PLoS ONE. 2011;6:e16103.
Muhlestein JB, Horne BD, Carlquist JF, et al. Cytomegalovirus seropositivity and C-reactive protein have independent and combined predictive value for mortality in patients with angiographically demonstrated coronary artery disease. Circulation. 2000;102:1917–23.
Naeger DM, Martin JN, Sinclair E, et al. Cytomegalovirus-specific T cells persist at very high levels during long-term antiretroviral treatment of HIV disease. PLoS One. 2010;5:e8886.
Gianella S, Morris SR, Tatro E, et al.: Virologic Correlates of Anti-CMV IgG Levels in HIV-1 Infected Men. J Infect Dis. 2013. doi:10.1093/infdis/jit434.
Parrinello CM, Sinclair E, Landay AL, et al. Cytomegalovirus immunoglobulin G antibody is associated with subclinical carotid artery disease among HIV-infected women. J Infect Dis. 2012;205:1788–96.
Hsue PY, Hunt PW, Sinclair E, et al. Increased carotid intima-media thickness in HIV patients is associated with increased cytomegalovirus-specific T-cell responses. AIDS. 2006;20:2275–83.
Durier N, Ananworanich J, Apornpong T, et al. Cytomegalovirus viremia in Thai HIV-infected patients on antiretroviral therapy: prevalence and associated mortality. Clin Infect Dis. 2013;57:147–55.
Hunt PW, Martin JN, Sinclair E, et al. Valganciclovir reduces T cell activation in HIV-infected individuals with incomplete CD4+ T cell recovery on antiretroviral therapy. J Infect Dis. 2011;203:1474–83.
van Baarle D, Tsegaye A, Miedema F, Akbar A. Significance of senescence for virus-specific memory T cell responses: rapid ageing during chronic stimulation of the immune system. Immunol Lett. 2005;97:19–29.
Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–83.
Deeks SG, Verdin E, McCune JM. Immunosenescence and HIV. Curr Opin Immunol. 2012;24:501–6.
Kaplan RC, Sinclair E, Landay AL, et al. T cell activation and senescence predict subclinical carotid artery disease in HIV-infected women. J Infect Dis. 2011;203:452–63.
Kaplan RC, Sinclair E, Landay AL, et al. T cell activation predicts carotid artery stiffness among HIV-infected women. Atherosclerosis. 2011;217:207–13.
Tincati C, Bellistri GM, Casana M, et al. CD8+ hyperactivation and senescence correlate with early carotid intima-media thickness in HIV+ patients with no cardiovascular disease. J Acquir Immune Defic Syndr. 2009;51:642–4.
Ford ES, Greenwald JH, Richterman AG, et al. Traditional risk factors and D-dimer predict incident cardiovascular disease events in chronic HIV infection. AIDS. 2010;24:1509–17.
Baker J, Huppler Hullsiek K, and Singh A, Monocyte Activation, but Not T Cell Activation, Predicts Progression of Coronary Artery Calcium in a Contemporary HIV Cohort (Abstract 66LB), in 20th Conference on Retroviruses and Opportunistic Infections 2013: Atlanta, GA, USA.
Tenorio A, Zheng E, and Bosch R, Soluble Markers of Inflammation and Coagulation, but Not T Cell Activation, Predict Non-AIDS-defining Events during Suppressive ART (Abstract 790), in 20th Conference on Retroviruses and Opportunistic Infections2013: Atlanta, GA, USA.
Unemori P, Leslie KS, Hunt PW, et al. Immunosenescence is associated with presence of Kaposi's sarcoma in antiretroviral treated HIV infection. AIDS. 2013;27:1735–42.
Lichtenstein K, Armon C, Buchacz K, et al. Low CD4+ T Cell Count Is a Risk Factor for Cardiovascular Disease Events in the HIV Outpatient Study. Clin Infect Dis. 2010;51:435–47.
Prosperi MC, Cozzi-Lepri A, Castagna A, et al. Incidence of malignancies in HIV-infected patients and prognostic role of current CD4 cell count: evidence from a large Italian cohort study. Clin Infect Dis. 2010;50:1316–21.
Onen NF, Agbebi A, Shacham E, et al. Frailty among HIV-infected persons in an urban outpatient care setting. J Infect. 2009;59:346–52.
Pathai S, Gilbert C, Weiss HA, et al. Frailty in HIV-infected adults in South Africa. J Acquir Immune Defic Syndr. 2013;62:43–51.
Yong MK, Elliott JH, Woolley IJ, Hoy JF. Low CD4 count is associated with an increased risk of fragility fracture in HIV-infected patients. J Acquir Immune Defic Syndr. 2011;57:205–10.
Guaraldi G, Orlando G, Zona S, et al. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin Infect Dis. 2011;53:1120–6.
Hasse B, Ledergerber B, Furrer H, et al. Morbidity and aging in HIV-infected persons: the Swiss HIV cohort study. Clin Infect Dis. 2011;53:1130–9.
Galli L, Salpietro S, Pellicciotta G, et al. Risk of type 2 diabetes among HIV-infected and healthy subjects in Italy. Eur J Epidemiol. 2012;27:657–65.
Malaza A, Mossong J, Barnighausen T, Newell ML. Hypertension and obesity in adults living in a high HIV prevalence rural area in South Africa. PLoS ONE. 2012;7:e47761.
Wand H, Ramjee G. High prevalence of obesity among women who enrolled in HIV prevention trials in KwaZulu-Natal, South Africa: healthy diet and life style messages should be integrated into HIV prevention programs. BMC Public Health. 2013;13:159.
Ramachandran A, Snehalatha C. Rising burden of obesity in Asia. J Obes. 2010(2010).
Tian S, Dong GH, Wang D, et al. Factors associated with prevalence, awareness, treatment and control of hypertension in urban adults from 33 communities in China: the CHPSNE Study. Hypertens Res. 2011;34:1087–92.
Kengne AP, Echouffo-Tcheugui JB, Sobngwi E, Mbanya JC. New insights on diabetes mellitus and obesity in Africa-part 1: prevalence, pathogenesis and comorbidities. Heart. 2013;99:979–83.
Hunter DJ, Reddy KS. Noncommunicable diseases. N Engl J Med. 2013;369:1336–43.
Tang S, Ehiri J, Long Q. China's biggest, most neglected health challenge: Non-communicable diseases. Infect Dis Poverty. 2013;2:7.
Hamill MM, Ward KA, Pettifor JM, Norris SA, and Prentice A: Bone mass, body composition and vitamin D status of ARV-naive, urban, black South African women with HIV infection, stratified by CD count. Osteoporos Int. 2013;24:2855–61.
Muronya W, Sanga E, Talama G, Kumwenda JJ, van Oosterhout JJ. Cardiovascular risk factors in adult Malawians on long-term antiretroviral therapy. Trans R Soc Trop Med Hyg. 2011;105:644–9.
Mateen FJ, Kanters S, Kalyesubula R, et al. Hypertension prevalence and Framingham risk score stratification in a large HIV-positive cohort in Uganda. J Hypertens. 2013;31:1372–8. discussion 1378.
Bloomfield GS, Hogan JW, Keter A, et al. Hypertension and obesity as cardiovascular risk factors among HIV seropositive patients in Western Kenya. PLoS ONE. 2011;6:e22288.
Tsai MS, Hung CC, Liu WC, et al. Reduced bone mineral density among HIV-infected patients in Taiwan: prevalence and associated factors. J Microbiol Immunol Infect. 2012. doi:10.1016/j.jmii.2012.08.026.
Erlandson KM, Allshouse AA, Jankowski CM, et al. Association of functional impairment with inflammation and immune activation in HIV type 1-infected adults receiving effective antiretroviral therapy. J Infect Dis. 2013;208:249–59.
Appay V, Fastenackels S, Katlama C, et al. Old age and anti-cytomegalovirus immunity are associated with altered T-cell reconstitution in HIV-1-infected patients. AIDS. 2011;25:1813–22.
Masia M, Robledano C. Ortiz de la Tabla V, et al.: Increased carotid intima-media thickness associated with antibody responses to varicella-zoster virus and cytomegalovirus in HIV-infected patients. PLoS ONE. 2013;8:e64327.
Cassol E, Malfeld S, Mahasha P, et al. Persistent microbial translocation and immune activation in HIV-1-infected South Africans receiving combination antiretroviral therapy. J Infect Dis. 2010;202:723–33.
Ledwaba L, Tavel JA, Khabo P, et al. Pre-ART levels of inflammation and coagulation markers are strong predictors of death in a South African cohort with advanced HIV disease. PLoS ONE. 2012;7:e24243.
Ford N, Shubber Z, Saranchuk P, et al.: Burden of HIV-Related Cytomegalovirus Retinitis in Resource-Limited Settings: A Systematic Review. Clin Infect Dis. 2013;57:1351–61.
Nakanjako D, Ssewanyana I, Nabatanzi R, et al. Impaired T-cell proliferation among HAART-treated adults with suboptimal CD4 recovery in an African cohort. BMC Immunol. 2013;14:26.
Hunt PW, Cao HL, Muzoora C, et al. Impact of CD8+ T-cell activation on CD4+ T-cell recovery and mortality in HIV-infected Ugandans initiating antiretroviral therapy. AIDS. 2011;25:2123–31.
Shiels PG, McGlynn LM, MacIntyre A, et al. Accelerated telomere attrition is associated with relative household income, diet and inflammation in the pSoBid cohort. PLoS ONE. 2011;6:e22521.
Atun R, Jaffar S, Nishtar S, et al. Improving responsiveness of health systems to non-communicable diseases. Lancet. 2013;381:690–7.
Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet. 2013;382:1525–33.
Nelson MR, Reid CM, Ames DA, et al. Feasibility of conducting a primary prevention trial of low-dose aspirin for major adverse cardiovascular events in older people in Australia: results from the ASPirin in Reducing Events in the Elderly (ASPREE) pilot study. Med J Aust. 2008;189:105–9.
O'Brien M, Montenont E, Hu L, et al. Aspirin attenuates platelet activation and immune activation in HIV-infected subjects on antiretroviral therapy: A Pilot Study. J Acquir Immune Defic Syndr. 2013;63:280–8. Reporting the striking finding of significantly reduced T cell actiavtion and sCD14 levels in virologically suppressed HIV+ individuals after only 1 week of treatment with aspirin. Although only a small study (25 HIV+ and 45 control participants), the data highlight the significant activation of platlets in HIV infection and the influence this has on monocyte activation and thus inflammation/immune activation. This is to date a poorly investigated area in HIV infection.
Antonopoulos AS, Margaritis M, Lee R, Channon K, Antoniades C. Statins as anti-inflammatory agents in atherogenesis: molecular mechanisms and lessons from the recent clinical trials. Curr Pharm Des. 2012;18:1519–30.
Calza L, Trapani F, Bartoletti M, et al. Statin Therapy Decreases Serum Levels of High-Sensitivity C-Reactive Protein and Tumor Necrosis Factor-α in HIV-Infected Patients Treated With Ritonavir-Boosted Protease Inhibitors. HIV Clinical Trials. 2012;13:153–61.
Baker JV, Huppler Hullsiek K, Prosser R, et al. Angiotensin Converting Enzyme Inhibitor and HMG-CoA Reductase Inhibitor as Adjunct Treatment for Persons with HIV Infection: A Feasibility Randomized Trial. PLoS ONE. 2012;7:e46894.
De Wit S, Delforge M, Necsoi CV, Clumeck N. Downregulation of CD38 activation markers by atorvastatin in HIV patients with undetectable viral load. Aids. 2011;25:1332–3.
Fichtenbaum CJ, Yeh TM, Evans SR, Aberg JA. Treatment with pravastatin and fenofibrate improves atherogenic lipid profiles but not inflammatory markers in ACTG 5087. J Clin Lipidol. 2010;4:279–87.
Rasmussen LD, Kronborg G, Larsen CS, et al. Statin Therapy and Mortality in HIV-Infected Individuals; A Danish Nationwide Population-Based Cohort Study. PLoS ONE. 2013;8:e52828.
Piconi S, Parisotto S, Rizzardini G, et al. Hydroxychloroquine drastically reduces immune activation in HIV-infected, antiretroviral therapy–treated immunologic nonresponders. Blood. 2011;118:3263–72.
Murray SM, Down CM, Boulware DR, et al. Reduction of immune activation with chloroquine therapy during chronic HIV infection. J Virol. 2010;84:12082–6.
Paton Ni GRLDDT et al. Effects of hydroxychloroquine on immune activation and disease progression among hiv-infected patients not receiving antiretroviral therapy: A randomized controlled trial. JAMA. 2012;308:353–61.
Cunningham-Rundles S, Ahrne S, Johann-Liang R, et al. Effect of probiotic bacteria on microbial host defense, growth, and immune function in human immunodeficiency virus type-1 infection. Nutrients. 2011;3:1042–70.
Klatt NR, Canary LA, Sun X, et al. Probiotic/prebiotic supplementation of antiretrovirals improves gastrointestinal immunity in SIV-infected macaques. J Clin Invest. 2013;123:903–7.
Gonzalez-Hernandez LA, Jave-Suarez LF, Fafutis-Morris M, et al. Synbiotic therapy decreases microbial translocation and inflammation and improves immunological status in HIV-infected patients: a double-blind randomized controlled pilot trial. Nutr J. 2012;11:90.
Schunter M, Chu H, Hayes TL, et al. Randomized pilot trial of a synbiotic dietary supplement in chronic HIV-1 infection. BMC Complement Alternat Med. 2012;12:84.
Lassenius MI, Pietilainen KH, Kaartinen K, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. 2011;34:1809–15.
Gonzalez-Quintela A, Alonso M, Campos J, et al. Determinants of serum concentrations of lipopolysaccharide-binding protein (LBP) in the adult population: the role of obesity. PLoS One. 2013;8:e54600.
De Luca A, de Gaetano DK, Colafigli M, et al. The association of high-sensitivity c-reactive protein and other biomarkers with cardiovascular disease in patients treated for HIV: a nested case–control study. BMC Infect Dis. 2013;13:414.
Koethe JR, Dee K, Bian A, et al. Circulating interleukin-6, soluble CD14, and other inflammation biomarker levels differ between obese and nonobese HIV-infected adults on antiretroviral therapy. AIDS Res Hum Retrovir. 2013;29:1019–25.
Hussein AA, Gottdiener JS, Bartz TM, et al. Inflammation and sudden cardiac death in a community-based population of older adults: The Cardiovascular Health Study. Heart Rhythm. 2013;10:1425–32.
Empana JP, Jouven X, Canoui-Poitrine F, et al. C-reactive protein, interleukin 6, fibrinogen and risk of sudden death in European middle-aged men: the PRIME study. Arterioscler Thromb Vasc Biol. 2010;30:2047–52.
Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–72.
Jenny NS, Tracy RP, Ogg MS, et al. In the elderly, interleukin-6 plasma levels and the -174G > C polymorphism are associated with the development of cardiovascular disease. Arterioscler Thromb Vasc Biol. 2002;22:2066–71.
Hsue PY, Scherzer R, Hunt PW, et al.: Carotid Intima-Media Thickness Progression in HIV-Infected Adults Occurs Preferentially at the Carotid Bifurcation and Is Predicted by Inflammation. Journal of the American Heart Association. 2012, 1. doi:10.1161/JAHA.111.000422.
Biron A, Bobin-Dubigeon C, Volteau C, et al. Metabolic syndrome in French HIV-infected patients: prevalence and predictive factors after 3 years of antiretroviral therapy. AIDS Res Hum Retrovir. 2012;28:1672–8.
Brown TT, Tassiopoulos K, Bosch RJ, Shikuma C, McComsey GA. Association between systemic inflammation and incident diabetes in HIV-infected patients after initiation of antiretroviral therapy. Diabetes Care. 2010;33:2244–9.
Schnabel RB, Yin X, Larson MG, et al. Multiple inflammatory biomarkers in relation to cardiovascular events and mortality in the community. Arterioscler Thromb Vasc Biol. 2013;33:1728–33.
Parkner T, Sorensen LP, Nielsen AR, et al. Soluble CD163: a biomarker linking macrophages and insulin resistance. Diabetologia. 2012;55:1856–62.
Hileman CO, Longenecker CT, Carman TL, et al. Elevated D-dimer is independently associated with endothelial dysfunction: a cross-sectional study in HIV-infected adults on antiretroviral therapy. Antivir Ther. 2012;17:1345–9.
Longenecker C, Funderburg N, Jiang Y, et al.: Markers of inflammation and CD8 T-cell activation, but not monocyte activation, are associated with subclinical carotid artery disease in HIV-infected individuals. HIV Med. 2013;14:385–90.
Merlini E, Luzi K, Suardi E, et al. T-cell phenotypes, apoptosis and inflammation in HIV+ patients on virologically effective cART with early atherosclerosis. PLoS One. 2012;7:e46073.
Shikuma CM, Barbour JD, Ndhlovu LC, et al.: Plasma Monocyte Chemoattractant Protein-1 and Tumor Necrosis Factor-alpha Levels Predict the Presence of Coronary Artery Calcium in HIV-Infected Individuals Independent of Traditional Cardiovascular Risk Factors. AIDS Res Hum Retroviruses. 2013. doi:10.1089/aid.2013.0183.
Aristoteli LP, Moller HJ, Bailey B, Moestrup SK, Kritharides L. The monocytic lineage specific soluble CD163 is a plasma marker of coronary atherosclerosis. Atherosclerosis. 2006;184:342–7.
Fjeldborg K, Christiansen T, Bennetzen M, et al. The Macrophage-Specific Serum Marker, Soluble CD163, Is Increased in Obesity and Reduced After Dietary-Induced Weight Loss. Obesity. 2013. doi:10.1002/oby.20376. Silver Spring.
Zanni MV, Burdo TH. Makimura H, Williams KC, and Grinspoon SK: Relationship between monocyte/macrophage activation marker soluble CD163 and insulin resistance in obese and normal-weight subjects. Clin Endocrinol (Oxf). 2012;77:385–90.
Moller HJ, Frikke-Schmidt R, Moestrup SK, Nordestgaard BG, Tybjaerg-Hansen A. Serum soluble CD163 predicts risk of type 2 diabetes in the general population. Clin Chem. 2011;57:291–7.
Vassallo M, Dunais B, Durant J, et al. Relevance of lipopolysaccharide levels in HIV-associated neurocognitive impairment: the Neuradapt study. J Neurovirol. 2013;19:376–82.
Weaver JD, Huang MH, Albert M, et al. Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology. 2002;59:371–8.
Noble JM, Manly JJ, Schupf N, et al. Association of C-reactive protein with cognitive impairment. Arch Neurol. 2010;67:87–92.
Ryan LA, Zheng J, Brester M, et al. Plasma levels of soluble CD14 and tumor necrosis factor-alpha type II receptor correlate with cognitive dysfunction during human immunodeficiency virus type 1 infection. J Infect Dis. 2001;184:699–706.
Kamat A, Lyons JL, Misra V, et al.: Monocyte Activation Markers in Cerebrospinal Fluid Associated With Impaired Neurocognitive Testing in Advanced HIV Infection. J Acquir Immune Defic Syndr 2012, 60:234–243 10.
Lyons JL, Uno H, Ancuta P, et al.: Plasma sCD14 Is a Biomarker Associated With Impaired Neurocognitive Test Performance in Attention and Learning Domains in HIV Infection. J Acquir Immune Defic Syndr 2011, 57:371–379 10.
Engelhart MJ, Geerlings MI, Meijer J, et al. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol. 2004;61:668–72.
Bruunsgaard H, Andersen-Ranberg K, Jeune B, et al. A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J Gerontol A Biol Sci Med Sci. 1999;54:M357–64.
Burdo TH, Weiffenbach A, Woods SP, et al. Elevated sCD163 in plasma but not cerebrospinal fluid is a marker of neurocognitive impairment in HIV infection. Aids. 2013;27:1387–95.
Blasko I, Knaus G, Weiss E, et al. Cognitive deterioration in Alzheimer's disease is accompanied by increase of plasma neopterin. J Psychiatr Res. 2007;41:694–701.
Marks MA, Rabkin CS, Engels EA, et al. Markers of microbial translocation and risk of AIDS-related lymphoma. AIDS. 2013;27:469–74.
Borges AH, Silverberg MJ, Wentworth D, et al. Predicting risk of cancer during HIV infection: the role of inflammatory and coagulation biomarkers. Aids. 2013;27:1433–41.
Il'yasova D, Colbert LH, Harris TB, et al. Circulating levels of inflammatory markers and cancer risk in the health aging and body composition cohort. Cancer Epidemiol Biomarkers Prev. 2005;14:2413–8.
Ding C, Parameswaran V, Udayan R, Burgess J, Jones G. Circulating levels of inflammatory markers predict change in bone mineral density and resorption in older adults: a longitudinal study. J Clin Endocrinol Metab. 2008;93:1952–8.
Scheidt-Nave C, Bismar H, Leidig-Bruckner G, et al. Serum interleukin 6 is a major predictor of bone loss in women specific to the first decade past menopause. J Clin Endocrinol Metab. 2001;86:2032–42.
Margolick J, Jacobson L, and Lopez J, Frailty and circulating concentrations of proinflammatory cytokines and chemokines in HIV-infected and -uninfected men in the Multicenter AIDS Cohort Study (MACS). , in 3rd International Workshop on HIV and Aging. 2012: Baltimore, USA.
Ferrucci L, Harris TB, Guralnik JM, et al. Serum IL-6 level and the development of disability in older persons. J Am Geriatr Soc. 1999;47:639–46.
Walston J, McBurnie MA, Newman A, et al. Frailty and activation of the inflammation and coagulation systems with and without clinical comorbidities: results from the Cardiovascular Health Study. Arch Intern Med. 2002;162:2333–41.
Leng SX, Tian X, Matteini A, et al. IL-6-independent association of elevated serum neopterin levels with prevalent frailty in community-dwelling older adults. Age Ageing. 2011;40:475–81.
Compliance with Ethics Guidelines
Conflict of Interest
Anna C. Hearps, Genevieve E. Martin, Reena Rajasuriar, and Suzanne M. Crowe declare that they have no conflict of interest
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hearps, A.C., Martin, G.E., Rajasuriar, R. et al. Inflammatory Co-morbidities in HIV+ Individuals: Learning Lessons from Healthy Ageing. Curr HIV/AIDS Rep 11, 20–34 (2014). https://doi.org/10.1007/s11904-013-0190-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11904-013-0190-8