
Abstract
Purpose of Review
To highlight recent results in targeting mRNA translation and discuss the results and prospects of translation inhibitors in cancer therapy.
Recent Findings
Until recently, inhibitors of mRNA translation have been thought to likely lack a therapeutic window. In 2012, the Food and Drug Administration (FDA) approved omacetaxine mepesuccinate (homoharringtonine) for the treatment of adults with chronic myelogenous leukemia (CML) who are resistant to at least two tyrosine kinase inhibitors. Since then, a few drugs, notably tomivosertib (eFT-508), selinexor (KPT-330), and ribavirin, have entered clinical trials. These drugs are known to inhibit mRNA translation. More recently, a number of interesting studies report that discrete subsets of proteins in cancer cells may be selectively targeted at the translation step, through inhibiting signals such as phospho-4E-BP1, eIF4A, and eIF4E. Promising therapies using these strategies have demonstrated potent anti-tumor activity in preclinical cancer models.
Summary
The growing number of translation inhibitors with diverse mechanisms, coupled with emerging insights into translational regulation of different cancer-promoting genes, suggests a bright new horizon for the field of therapeutic targeting of mRNA translation in cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is characterized by unchecked cellular growth and requires a high level of mRNA translation. Translation can be divided into three stages: translational initiation, elongation, and termination. A growing number of translation inhibitors have been developed to inhibit mainly translational initiation or elongation (Fig. 1). Translational initiation involves various kinases that stimulate phosphorylation of eukaryotic initiation factor 4E (eIF4E)–binding proteins (4E-BP) such as 4E-BP1. In its hypo-phosphorylated state, 4E-BP1 sequesters eIF4E and acts as a “brake” for initiation of translation [1]. mTORC1 has been established as a key activator for hyper-phosphorylation of 4E-BP1 and translational initiation [2,3,4]. Casein kinase 1 epsilon (CK1ε) and other kinases have been implicated in stimulating hyper-phosphorylation of 4E-BP1 [5, 6••]. The release of eIF4E by hyper-phosphorylation of 4E-BP1 is followed by assembly of the eukaryotic initiation factor 4F (eIF4F), which is a critical event for cap-dependent translation. eIF4F comprises 3 subunits, i.e., the mRNA 5′-cap-binding subunit eIF4E, the large scaffolding subunit eIF4G, and the RNA helicase subunit eIF4A (Fig. 1). Subsequently, the 40S and 60S ribosomes are assembled on the AUG codon of the mRNA, thus starting the translation.

Description of mRNA translation. Initiation and elongation are the two main steps of the three-phased cap-dependent translation machinery. Initiation, a rate-limiting step, involves the interaction of key initiation factors eIF4A, eIF4E, and eIF4G with the cap structure at the 5′ end of the mRNA as well as the 40S ribosomal subunit and assembly into the eIF4F complex. The assembled complex then scans the mRNA to find the start codon (AUG) where the 40S and 60S ribosome subunits are assembled onto the mRNA. Elongation starts at the end of the initiation step where ribosomes construct amino acids into polypeptides and protein synthesis begins
While the process of cap-dependent translation depicted in Fig. 1 is shared among the vast majority of proteins, except for those with a highly active internal ribosome entry site (IRES), there is a wide range of protein abundance and susceptibility to translation inhibitors. Protein levels are regulated at transcriptional, translational, and post-translational levels. Proteins with short half-lives, including many oncogenic proteins, may be particularly affected by translation inhibitors. For oncoproteins that are essential for cancer cell survival and proliferation, targeting mRNA translation can be a very productive therapeutic strategy. For example, the CML driver oncogene BCR-ABL is highly prone to translational inhibition by omacetaxine mepesuccinate, underpinning the success of the drug in treating CML. Omacetaxine is an FDA-approved natural product that directly inhibits mRNA translation.
The MYC oncogene is overexpressed in many human cancers [7] and has a half-life of only 30 min [8]. Dysregulation of the MYC oncogene, in the form of protein overexpression or gene translocation, is observed in 30–40% of all diffuse large B cell lymphomas (DLBCL), and these constitute the majority of chemo-resistant and incurable DLBCL [9,10,11,12,13,14,15]. Dysregulation of C-MYC is also common in multiple myeloma and is associated with inferior clinical outcome [16,17,18,19]. Similarly, MYC activation is observed in 40% of pancreatic ductal adenocarcinoma cases (PDAC) [20, 21], and dysregulated C-MYC is associated with high-risk PDAC and shortened survival [22, 23]. Despite the obvious importance of targeting C-MYC, direct inhibitors have not been successfully developed. The natural products silvestrol and pateamine [24,25,26,27,28], which inhibit the function of eIF4A and thus interfere with mRNA translation, have been shown to downregulate C-MYC. Interestingly, we recently discovered that umbralisib (TGR-1202) and carfilzomib, two clinically available drugs that are seemingly unrelated, form a highly synergistic combination to inhibit eIF4F-dependent translation of C-MYC [6••]. Furthermore, the synergy of umbralisib and carfilzomib is driven in part by the ability of umbralisib to inhibit CK1ε in addition to its known target phosphoinositide 3-kinase delta (PI3Kδ). The above results suggest that translation inhibitors could contribute to tumor regression by preventing oncogene-enabled cancer cell proliferation and survival.
Recent data suggest that translation inhibitors may also have a therapeutic role in activating an innate anti-tumor immune response, through repressing the expression of programmed death-ligand 1 (PD-L1/CD274) [29••, 30••]. In a double transgenic mouse model of liver cancer, tumorigenesis was driven by both the activating KRAS(G12D) mutation and overexpression of C-MYC [29••]. The liver cancer cells demonstrated constitutively activated translation of PD-L1, leading to evasion of innate anti-tumor surveillance and eradication. eFT508, which inhibits eIF4E phosphorylation, was shown to potently inhibit translation of PD-L1 and reverse the aggressive and metastatic characteristics of the liver cancer driven by KRAS(G12D) and C-MYC. In a mouse model of melanoma, STAT1-mediated transcription of PD-L1 was induced by interferon gamma via upregulation of STAT1 translation [30••]. Interestingly, the translation of STAT1 could be inhibited by silvestrol, pateamine, and siRNA targeting eIF4A or eIF4E. Consistent with the induction of innate immunity, silvestrol effectively treated melanoma in immune-competent mice, but not in immune-compromised mice such as nude mice and CD8+ T cell–depleted mice.
The above results demonstrate that various translation inhibitors can be used with acceptable safety to produce robust anti-tumor effects in animals and in humans. Conceptually, translation inhibitors may preferentially inhibit cancer cells while preserving normal cells to a degree through two related or overlapping mechanisms. First, cancer cells may be more sensitive to repression of global translation. Such precedence is widely observed with DNA- or microtubule-targeting drugs like doxorubicin and paclitaxel, respectively. Although these drugs do not necessarily differentiate the macromolecular targets in cancer versus normal cells, these drugs are nevertheless effective and safe for the treatment of a wide variety of malignancies. Secondly, many cancer-promoting genes may be highly expressed and critically needed in cancer but not normal cells, and the translation of these genes may be more susceptible to translational inhibition than other genes. CMLD010509, a member of the rocaglate family that also includes silvestrol, is reported to cause depletion of short-lived proteins including oncoproteins like C-MYC, MDM2, CCND1, MAF, and MCL-1. CMLD010509 has been shown to be safe and active in mouse models of myeloma. It is conceivable that targeting multiple cancer-promoting genes using translation inhibitors may be highly desirable, because such treatments may overcome inherent resistance of the cancer cells that possess complex and compensatory oncogenic signals. On the other hand, in the rare case of a malignancy that is addicted to a single gene, such as BCL-ABL in CML, translation inhibitors may be highly effective and safe.
Two broad strategies can be envisioned to improve the precision and therapeutic window of targeting protein translation in cancer. The first approach could focus on identifying translational regulators that are preferentially activated or overexpressed in a subset of tumors but not in other tumors or normal tissues. Based on the select studies discussed above, it is highly possible that tumors that possess overexpressed or activated eIF4E, mTORC1, and Mnk1 may be selectively more sensitive to translation inhibitors, compared with normal tissues and tumors without such pathological markers. The second approach may involve identifying translationally regulated genes that are overexpressed or activated in cancer but not normal tissues. As discussed above, C-MYC overexpression is widespread in DLBCL and other lymphomas. An elegant study using ribosome footprinting reveals that C-MYC and a number of other oncogenes possess 5′ untranslated region (UTR) sequences that can form RNA G-quadruplex structure [24]. Such a structure poses a barrier for eIF4A to initiate translation, and genes with this structure, such as C-MYC and CCND3, are more susceptible to repression by the eIF4A inhibitor silvestrol. These results suggest that cancers that are dependent on C-MYC or other oncogenes with RNA G-quadruplex structures may be the preferred cancers for treatments such as silvestrol. Highly sophisticated studies have been generating unprecedented insights into the translation of thousands of genes at the same time [25, 31,32,33,34,35]. Such insights into single-gene translation, combined with ever-increasing amount of data from tumor sequencing efforts, will accelerate the clinical development of translation inhibitors in cancer.
In the following section, we review the therapeutic targets and promising experimental drugs that may be used to target protein translation in cancer.
-
1.
Targeting 4E-BP1 using umbralisib (TGR-1202) and carfilzomib. Our group recently discovered that TGR-1202 (umbralisib) and carfilzomib form a highly synergistic combination in DLBCL [6••]. We discovered that TGR-1202 is a first-in-class dual inhibitor of PI3Kδ and CK1ε. Carfilzomib is a proteasome inhibitor, which is approved for multiple myeloma but has no clinical activity in lymphoma. Combining the two drugs at clinically achievable concentration induces marked apoptosis in lymphoma cell lines and fresh primary lymphoma cells in vitro. Mechanistically, the two drugs synergize to inhibit phosphorylation of 4E-BP1 and translation of C-MYC, and the synergy is overcome by overexpressing eIF4E or C-MYC. The two drugs are now being studied in a phase I clinical trial (NCT02867618).
-
2.
Targeting 4E-BP1 using mTOR inhibitors. mTORC1 is a well-characterized activator causing hyper-phosphorylation of 4E-BP1, leading to the release of eIF4E from 4E-BP1, assembly of the eIF4F complex, and robust mRNA translation [2,3,4]. In keeping with these data, mTORC1 and dual mTORC1/mTORC2 inhibitors have been found to cause varying degrees of inhibition of 4E-BP1 hyper-phosphorylation and translation initiation for tumor-promoting genes [36,37,38,39,40,41,42]. However, the therapeutic effects of mTOR inhibition in cancer remain poorly understood. mTORC1 inhibitors such as everolimus and temsirolimus have been approved for renal cell cancer (RCC), but they demonstrate limited activity in other cancers such as DLBCL. The dual mTORC1/mTORC2 inhibitor MLN0128 was recently reported to exhibit no activity in lymphoma [39].
-
3.
Targeting eIF4A using pateamine and silvestrol/rocaglate analogs. Silvestrol is a rocaglate derivative isolated from the plant Aglaia foveolata [43]. It binds to the eIF4A subunit and increases its mRNA-binding activity, thereby limiting its participation in eIF4F complex formation [44, 45]. Silvestrol has been studied extensively and its anti-tumor activity has been demonstrated both in vitro and in vivo in several cancer models including melanoma, breast and prostate cancers, chronic lymphocytic leukemia (CLL) and acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), hepatocellular cancers (HCC), and brain cancer (meningioma) [44,45,46,47,48,49,50,51]. All demonstrate efficacy of this molecule at nanomolar concentrations in tumor cell lines, reporting antiproliferative activity, induction of apoptosis, G2/M cell cycle arrest, and decreased expression of important oncogenic/signaling proteins such as C-MYC, MDM2, MYB, NOTCH-1, ETS1, PCNA, AKT, ERK, MCL-1, BCL-2, AND BCL-XL, and cyclins D1, D3, E, and A, with different reductions occurring in different cellular contexts (e.g., different cell lines and tumor types). Additionally, studies have also demonstrated the anti-tumor effects of silvestrol in xenograft models improving survival with the compound being well tolerated in animals [24, 44,45,46,47,48,49,50,51]. Synergistic effects of silvestrol combined with other chemotherapy agents such as daunorubicin, etoposide, cytarabine, cisplatin, sorafenib, and rapamycin have also been demonstrated in several studies [49, 52, 53]. It has been shown to be a substrate and inducer of the multi-drug resistance 1 (MDR1/ABCB1) efflux pump [54], which may limit silvestrol activity as well as its absorption in the GI tract.
The class of Pateamine A (PatA)–derived compounds are natural compounds originally isolated from the marine sponge Mycale sp. [55]. PatA inhibits translation initiation through binding to eIF4A [26, 47, 56]. PatA was initially developed as an immunosuppressive agent inhibiting T cell receptor–induced IL-2 production [57, 58]. Later it was shown to inhibit cell growth and induce apoptosis in several cell lines [26, 59]. Des-methyl des-amino PatA (DMDAPatA) is a synthetic analog of PatA with more potent in vitro activity than the natural product PatA [58, 60]. The in vitro anti-tumor activity of DMDAPatA has been demonstrated in several cancer cell lines [28, 59, 61, 62]. Further, its anti-tumor effect in several nude mice xenograft models displayed significant inhibition of tumor growth in melanoma models albeit modest activity in non-small cell lung cancer and colon cancer cell lines, but without significant effect on pancreatic and HT-29 colon cancer xenograft models [62]. DMDAPatA and its analogs reduce the level of C-MYC, cyclin D1, MDM2, and MCl-1 proteins and have been shown to induce S-phase arrest in several cancer cell lines [28, 61, 62]. Unlike silvestrol, DMDAPatA is not a substrate for MDR1-mediated efflux [62]. However, potency may be limited by high-level DMDAPatA binding to human plasma proteins (> 99%). As a result, new analogs of this compound with less plasma protein binding, more potency, and greater selectivity towards cancer cells have been developed [61].
-
4.
Targeting eIF4E using the Mnk1/Mnk2 inhibitor eFT508. The eukaryotic initiation factor eIF4E plays a significant role in cap-dependent translation and is regulated by the mitogen-activated protein kinase-interacting kinases 1/2 (Mnk1/2) [63, 64]. MNK1 and MNK2 incorporate signals from different oncogenic and immunogenic stimuli such as RAS, p38, and Toll-like receptor (TLR) and regulate the translation of several subsets of mRNAs by phosphorylation of eIF4E [65, 66]. eFT508 is a potent, highly selective, and dual MNK1/2 inhibitor [67] and now in phase 2 clinical trials in lymphoma (NCT02937675) and in advanced solid tumors (NCT02605083). Recently, Xu et al. demonstrated that eFT508 produced significant and selective downregulation of PD-L1 translation driven by Kras and c-Myc, and the drug is effective in the transgenic mouse model of hepatic cell cancer [29••]. Based on this observation, a phase 2 clinical trial in relapsed or refractory microsatellite stable colorectal cancer (MSS CRC) is being conducted combining eFT508 and avelumab an anti-PD-L1 checkpoint inhibitor (NCT03258398).
-
5.
Targeting eIF4E using ribavirin. Ribavirin is an FDA-approved antiviral agent used for the treatment of respiratory syncytial virus and hepatitis C [68]. Ribavirin binds directly to eIF4E with micromolar affinity and competitively inhibits eIF4E:m7G mRNA cap-binding and disrupts eIF4E-mediated nuclear to cytoplasmic mRNA export [69]. Pharmacologic inhibition of eIF4E with ribavirin inhibits proliferation and survival of leukemic blasts derived from infants with acute lymphoblastic leukemia [70]. Ribavirin showed promising outcomes in relapsed and refractory AML [71] in combination with cytarabine [72] and is being evaluated in follicular and mantle cell lymphoma (NCT03585725). A clinically achievable concentration of ribavirin significantly inhibits chronic lymphocytic leukemia (CLL) lymphocytes in vitro with fludarabine [73]. Ribavirin has demonstrated clinical activity in solid tumors such as breast cancer (NCT01056757), oropharyngeal cancer (NCT01721525), prostate cancer, and other solid tumors (NCT01309490) [74, 75].
-
6.
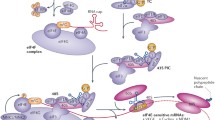
Targeting eIF4E-associated nuclear export of mRNA using selinexor. The karyopherin family of proteins are a large group of transporters that are responsible for the import (importins) and export (exportins) of proteins and other molecules into and out of the nucleus. They recognize proteins labeled with the nuclear localization signal (NLS) or nuclear export sequence (NES) and facilitate their import and export into and out of the nucleus, respectively. XPO1 (CRM1) is the best-characterized exportin transporters that recognize the NES of cargo proteins (including tumor suppressors like p53, p21, BRCA1/2, cell cycle regulatory proteins, transcription and translation factors, and miRNAs, which are frequently upregulated in cancers) and facilitates their export from nucleus to the cytoplasm (Fig. 2) [76,77,78]. Additionally, XPO1 facilitates transport to the cytoplasm the mRNAs that possess a 50-nucleotide element in the 3′ untranslated region denoted as the eIF4E sensitivity element, such as c-Myc, cyclin D1, and MDM2 (Fig. 2) [79,80,81].

Nuclear export of mRNA and action of selinexor. While eIF4E is mainly involved in translational initiation in the cytoplasm, eIF4E also acts in the nucleus where it promotes the export of a specific subset of mRNAs that contain a 50-nucleotide element in the 3′ untranslated region denoted as an eIF4E sensitivity element. Xpo1 is an exportin transporter that forms an export complex with eIF4E in the nucleus and facilitates the transport of eIF4E target mRNAs from the nucleus into the cytoplasm where they undergo cap-dependent translation. The hydrolysis of the Ran-GTP enables the release of Xpo1 cargo in the cytoplasm. Selinexor targets the Xpo1 protein recognition site and inhibits its nuclear export activity
Selinexor (KPT-330) (Fig. 2) is a selective inhibitor of nuclear export (SINE) that targets the cysteine 528 residue of XPO1 essential for NES recognition. KPT-330 has also been reported to transiently degrade the XPO1 protein, which is upregulated in many types of cancers [76, 82,83,84]. Further, selinexor blocks XPO1-mediated nuclear export of tumor suppressors including p53, p21, and BRCA1/2; growth regulatory proteins such as MYC and BCR-ABL; transcription and translation factors; and miRNAs [83, 84]. Several preclinical studies have demonstrated the ability of selinexor to inhibit growth and induce apoptosis in different cancer models, many of them though the restoration of tumor suppressor proteins [85,86,87,88,89]. Selinexor-mediated downregulation of the cap-dependent translation of several oncogenes including MYC, CDC25A, BCL-2, BCL-XL, and MCL-1 has been reported [86]. Selinexor has been and is currently being examined in over 60 clinical trials of various types of hematological and solid tumor cancers, alone or in combination with other chemotherapy agents [90]. It has demonstrated promising outcomes in acute myeloid leukemia [91, 92], advanced refractory bone or soft tissue sarcomas [93], and refractory multiple myeloma for which it has received orphan drug designation from the US FDA [94]. Other SINEs such as verdinexor (KPT-335) have also been developed; however, selinexor remains the most promising yet for cancer treatment.
-
7.
Targeting translational elongation using homoharringtonine. Omacetaxine mepesuccinate, a semisynthetic analog of homoharringtonine, is approved for third-line treatment of CML [95, 96]. Homoharringtonine (omacetaxine) prevents the elongation step of protein synthesis by interacting with the A-site of the ribosome and disrupting the positioning of aminoacyl-tRNAs [97,98,99]. Outside of its approved indication in CML, omacetaxine has been studied in various combination regimens for AML. In a phase 2 clinical study, homoharringtonine and sorafenib in combination have demonstrated encouraging activity in relapsed or refractory FLT3-ITD AML [100]. Homoharringtonine has been safely combined with cytarabine for the treatment of AML, resulting in the improvement of complete remission [101]. In a randomized, controlled, phase 3 study of 620 patients with de novo AML, the triple-drug regimen of homoharringtonine, cytarabine, and daunorubicin substantially outperforms the combination of cytarabine and daunorubicin [102].
Conclusion
Globally, cancer is responsible for about 9.6 million deaths annually. New targets and concepts are urgently needed to address cancer-related mortality and morbidity. While translation inhibitors have gradually entered clinical development, new insights into how these agents preferentially repress the level of key proteins, such as C-MYC and PD-L1, could substantially accelerate the rate of discovery and clinical development. Work on predictive biomarkers is critically needed to treat the right patient population.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Haghighat A, Mader S, Pause A, Sonenberg N. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 1995;14:5701–9.
Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, et al. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 1999;13:1422–37.
Hara K, Yonezawa K, Kozlowski MT, Sugimoto T, Andrabi K, Weng QP, et al. Regulation of eIF-4E BP1 phosphorylation by mTOR. J Biol Chem. 1997;272:26457–63.
Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–87.
Shin S, Wolgamott L, Roux PP, Yoon SO. Casein kinase 1epsilon promotes cell proliferation by regulating mRNA translation. Cancer Res. 2014;74:201–11.
•• Deng C, Lipstein MR, Scotto L, Jirau Serrano XO, Mangone MA, Li S, et al. Silencing c-Myc translation as a therapeutic strategy through targeting PI3Kdelta and CK1epsilon in hematological malignancies. Blood. 2017;129:88–99 The results demonstrate that clinically available drugs, for example, umbralisib and carfilzomib, can be combined in rational combinations to synergistically inhibit translation. Potentially many other combinations can be identified to silence translation, thus avoiding the delays in developing brand new translation inhibitors of uncertain clinical value.
Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. 2013;3.
Andresen C, Helander S, Lemak A, Farès C, Csizmok V, Carlsson J, et al. Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res. 2012;40:6353–66.
Savage KJ, Johnson NA, Ben-Neriah S, Connors JM, Sehn LH, Farinha P, et al. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood. 2009;114:3533–7.
Barrans S, Crouch S, Smith A, Turner K, Owen R, Patmore R, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol. 2010;28:3360–5.
Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S, et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012;30:3452–9.
Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood. 2013;121:4021–31.
Green TM, Young KH, Visco C, Xu-Monette ZY, Orazi A, Go RS, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012;30:3460–7.
Lin P, Medeiros LJ. High-grade B-cell lymphoma/leukemia associated with t(14;18) and 8q24/MYC rearrangement: a neoplasm of germinal center immunophenotype with poor prognosis. Haematologica. 2007;92:1297–301.
Copie-Bergman C, Cuilliere-Dartigues P, Baia M, Briere J, Delarue R, Canioni D, et al. MYC-IG rearrangements are negative predictors of survival in DLBCL patients treated with immunochemotherapy: a GELA/LYSA study. Blood. 2015;126:2466–74.
Carrasco DR, Tonon G, Huang Y, Zhang Y, Sinha R, Feng B, et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell. 2006;9:313–25.
Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia. 2014;28:1725–35.
Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. 2015;6:6997.
Chng WJ, Huang GF, Chung TH, Ng SB, Gonzalez-Paz N, Troska-Price T, et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia. 2011;25:1026–35.
Schleger, C., Verbeke, C., Hildenbrand, R., Zentgraf, H. & Bleyl, U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol 15, 462–469 (2002).
Hessmann E, Schneider G, Ellenrieder V, Siveke JT. MYC in pancreatic cancer: novel mechanistic insights and their translation into therapeutic strategies. Oncogene. 2016;35:1609–18.
Chen R, Dawson DW, Pan S, Ottenhof NA, de Wilde RF, Wolfgang CL, et al. Proteins associated with pancreatic cancer survival in patients with resectable pancreatic ductal adenocarcinoma. Lab Investig. 2015;95:43–55.
Zhang M, et al. Three new pancreatic cancer susceptibility signals identified on chromosomes 1q32.1, 5p15.33 and 8q24.21. Oncotarget. 2016.
Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature. 2014;513:65–70.
Iwasaki S, Floor SN, Ingolia NT. Rocaglates convert DEAD-box protein eIF4A into a sequence-selective translational repressor. Nature. 2016;534:558–61.
Low WK, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick WC, et al. Inhibition of eukaryotic translation initiation by the marine natural product pateamine a. Mol Cell. 2005;20:709–22.
Manier S, et al. Inhibiting the oncogenic translation program is an effective therapeutic strategy in multiple myeloma. Sci Transl Med. 2017;9.
Kim YR, et al. Silencing oncogene translation using pateamine a analogues as a novel therapeutic strategy for c-Myc driven lymphoma. Blood. 2017;130:–4111.
•• Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med. 2019;25:301–11 This paper demonstrates that eFT508, which is now in early phase clinical trials, can preferentially inhibit translation of PD-L1 and induce tumor regression in animal models. This is a significant step forward in establishing that targeting translation can invoke two mechanisms to control tumor, by directly inducing apoptosis and indirectly stimulating the anti-tumor immune response.
•• Cerezo M, Guemiri R, Druillennec S, Girault I, Malka-Mahieu H, Shen S, et al. Translational control of tumor immune escape via the eIF4F-STAT1-PD-L1 axis in melanoma. Nat Med. 2018;24:1877–86 This paper is the first to convincingly demonstrate that targeting eIF4F can preferentially inhibit translation of STAT1, leading to reduced transcription of PD-L1 and stimulation of anti-tumor immune response.
Ingolia NT. Ribosome footprint profiling of translation throughout the genome. Cell. 2016;165:22–33.
Gonzalez C, Sims JS, Hornstein N, Mela A, Garcia F, Lei L, et al. Ribosome profiling reveals a cell-type-specific translational landscape in brain tumors. J Neurosci. 2014;34:10924–36.
Simsek D, et al. The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell. 2017;169:1051–1065 e18.
Leppek K, Das R, Barna M. Functional 5' UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol. 2018;19:158–74.
Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, et al. Differential requirements for eIF4E dose in normal development and cancer. Cell. 2015;162:59–71.
Yun S, Vincelette ND, Knorr KLB, Almada LL, Schneider PA, Peterson KL, et al. 4EBP1/c-MYC/PUMA and NFkappaB/EGR1/BIM pathways underlie cytotoxicity of mTOR dual inhibitors in malignant lymphoid cells. Blood. 2016;127:2711–22.
Zhang C, et al. Icariside II, a natural mTOR inhibitor, disrupts aberrant energy homeostasis via suppressing mTORC1-4E-BP1 axis in sarcoma cells. Oncotarget. 2016.
Demosthenous C, Han JJ, Stenson MJ, Maurer MJ, Wellik LE, Link B, et al. Translation initiation complex eIF4F is a therapeutic target for dual mTOR kinase inhibitors in non-Hodgkin lymphoma. Oncotarget. 2015;6:9488–501.
Ghobrial IM, Siegel DS, Vij R, Berdeja JG, Richardson PG, Neuwirth R, et al. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: a phase I dose escalation study in patients with relapsed or refractory multiple myeloma, non-Hodgkin lymphoma, or Waldenstrom’s macroglobulinemia. Am J Hematol. 2016;91:400–5.
Kuo SH, Hsu CH, Chen LT, Lu YS, Lin CH, Yeh PY, et al. Lack of compensatory pAKT activation and eIF4E phosphorylation of lymphoma cells towards mTOR inhibitor, RAD001. Eur J Cancer. 2011;47:1244–57.
O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8.
Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61.
Hwang BY, Su BN, Chai H, Mi Q, Kardono LB, Afriastini JJ, et al. Silvestrol and episilvestrol, potential anticancer rocaglate derivatives from Aglaia silvestris. J Org Chem. 2004;69:3350–8.
Bordeleau ME, et al. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest. 2008;118.
Cencic R, et al. Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS One. 2009;4.
Lucas DM, et al. The novel plant-derived agent silvestrol has B-cell selective activity in chronic lymphocytic leukemia and acute lymphoblastic leukemia in vitro and in vivo. Blood. 2009;113.
Babendure JR, Babendure JL, Ding JH, Tsien RY. Control of mammalian translation by mRNA structure near caps. RNA. 2006;12.
Chen W-L, Pan L, Kinghorn AD, Swanson SM, Burdette JE. Silvestrol induces early autophagy and apoptosis in human melanoma cells. BMC Cancer. 2016;16:17.
Kogure T, Kinghorn AD, Yan I, Bolon B, Lucas DM, Grever MR, et al. Therapeutic potential of the translation inhibitor silvestrol in hepatocellular cancer. PLoS One. 2013;8:e76136.
Oblinger JL, Burns SS, Huang J, Pan L, Ren Y, Shen R, et al. Overexpression of eIF4F components in meningiomas and suppression of meningioma cell growth by inhibiting translation initiation. Exp Neurol. 2018;299:299–307.
Kim S, et al. Silvestrol, a potential anticancer rocaglate derivative from Aglaia foveolata, induces apoptosis in LNCaP cells through the mitochondrial/apoptosome pathway without activation of executioner caspase-3 or -7. Anticancer Res. 2007;27:2175–83.
Cencic R, Carrier M, Trnkus A, Porco JA Jr, Minden M, Pelletier J. Synergistic effect of inhibiting translation initiation in combination with cytotoxic agents in acute myelogenous leukemia cells. Leuk Res. 2010;34:535–41.
Daker M, et al. Inhibition of nasopharyngeal carcinoma cell proliferation and synergism of cisplatin with silvestrol and episilvestrol isolated from Aglaia stellatopilosa. Exp Ther Med. 2016;11:2117–26.
Gupta SV, Sass EJ, Davis ME, Edwards RB, Lozanski G, Heerema NA, et al. Resistance to the translation initiation inhibitor silvestrol is mediated by ABCB1/P-glycoprotein overexpression in acute lymphoblastic leukemia cells. AAPS J. 2011;13:357–64.
Northcote PT, Blunt JW, Munro MHG. Pateamine: a potent cytotoxin from the New Zealand marine sponge, Mycale sp. Tetrahedron Lett. 1991;32:6411–4.
Low WK, et al. Isolation and identification of eukaryotic initiation factor 4A as a molecular target for the marine natural product pateamine A. Methods Enzymol. 2007;431:303–24 (Academic Press.
Romo D, Rzasa RM, Shea HA, Park K, Langenhan JM, Sun L, et al. Total synthesis and immunosuppressive activity of (−)-pateamine A and related compounds: implementation of a β-lactam-based macrocyclization. J Am Chem Soc. 1998;120:12237–54.
Romo D, Choi NS, Li S, Buchler I, Shi Z, Liu JO. Evidence for separate binding and scaffolding domains in the immunosuppressive and antitumor marine natural product, pateamine a: design, synthesis, and activity studies leading to a potent simplified derivative. J Am Chem Soc. 2004;126:10582–8.
Hood KA, West LM, Northcote PT, Berridge MV, Miller JH. Induction of apoptosis by the marine sponge (Mycale) metabolites, mycalamide A and pateamine. Apoptosis. 2001;6:207–19.
Low W-K, Li J, Zhu M, Kommaraju SS, Shah-Mittal J, Hull K, et al. Second-generation derivatives of the eukaryotic translation initiation inhibitor pateamine A targeting eIF4A as potential anticancer agents. Bioorg Med Chem. 2014;22:116–25.
Chen R, Zhu M, Chaudhari RR, Robles O, Chen Y, Skillern W, et al. Creating novel translation inhibitors to target pro-survival proteins in chronic lymphocytic leukemia. Leukemia. 2019.
Kuznetsov G, Xu Q, Rudolph-Owen L, TenDyke K, Liu J, Towle M, et al. Potent in vitro and in vivo anticancer activities of des-methyl, des-amino pateamine A, a synthetic analogue of marine natural product pateamine A. Mol Cancer Ther. 2009;8:1250–60.
Korneeva NL, Song A, Gram H, Edens MA, Rhoads RE. Inhibition of mitogen-activated protein kinase (MAPK)-interacting kinase (MNK) preferentially affects translation of mRNAs containing both a 5′-terminal cap and hairpin. J Biol Chem. 2016;291:3455–67.
Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol. 2004;24:6539–49.
Joshi S, Platanias LC. Mnk kinases in cytokine signaling and regulation of cytokine responses. Biomol Concepts. 2012;3:127–39.
Joshi S, Platanias LC. Mnk kinase pathway: cellular functions and biological outcomes. World J Biol Chem. 2014;5:321–33.
Reich SH, Sprengeler PA, Chiang GG, Appleman JR, Chen J, Clarine J, et al. Structure-based design of pyridone-aminal eFT508 targeting dysregulated translation by selective mitogen-activated protein kinase interacting kinases 1 and 2 (MNK1/2) inhibition. J Med Chem. 2018;61:3516–40.
Riner A, Chan-Tack KM, Murray JS. Original research: intravenous ribavirin--review of the FDA’s emergency investigational new drug database (1997-2008) and literature review. Postgrad Med. 2009;121:139–46.
Borden KL, Culjkovic-Kraljacic B. Ribavirin as an anti-cancer therapy: acute myeloid leukemia and beyond? Leuk Lymphoma. 2010;51:1805–15.
Urtishak KA, Wang LS, Culjkovic-Kraljacic B, Davenport JW, Porazzi P, Vincent TL, et al. Targeting EIF4E signaling with ribavirin in infant acute lymphoblastic leukemia. Oncogene. 2019;38:2241–62.
Assouline S, Culjkovic B, Cocolakis E, Rousseau C, Beslu N, Amri A, et al. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): a proof-of-principle clinical trial with ribavirin. Blood. 2009;114:257–60.
Kraljacic BC, Arguello M, Amri A, Cormack G, Borden K. Inhibition of eIF4E with ribavirin cooperates with common chemotherapies in primary acute myeloid leukemia specimens. Leukemia. 2011;25:1197–200.
Martinez-Marignac V, Shawi M, Pinedo-Carpio E, Wang X, Panasci L, Miller W, et al. Pharmacological targeting of eIF4E in primary CLL lymphocytes. Blood Cancer J. 2013;3:e146.
Dunn LA, Fury MG, Sherman EJ, Ho AA, Katabi N, Haque SS, et al. Phase I study of induction chemotherapy with afatinib, ribavirin, and weekly carboplatin and paclitaxel for stage IVA/IVB human papillomavirus-associated oropharyngeal squamous cell cancer. Head Neck. 2018;40:233–41.
Kosaka T, Nagamatsu G, Saito S, Oya M, Suda T, Horimoto K. Identification of drug candidate against prostate cancer from the aspect of somatic cell reprogramming. Cancer Sci. 2013;104:1017–26.
Jans DA, Martin AJ, Wagstaff KM. Inhibitors of nuclear transport. Curr Opin Cell Biol. 2019;58:50–60.
Çağatay T, Chook YM. Karyopherins in cancer. Curr Opin Cell Biol. 2018;52:30–42.
Xu D, Grishin NV, Chook YM. NESdb: a database of NES-containing CRM1 cargoes. Mol Biol Cell. 2012;23:3673–6.
Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KLB. eIF4E promotes nuclear export of cyclin D1 mRNAs via an element in the 3′UTR. J Cell Biol. 2005;169:245–56.
Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KL. eIF4E is a central node of an RNA regulon that governs cellular proliferation. J Cell Biol. 2006;175:415–26.
Culjkovic-Kraljacic, B., Baguet, A., Volpon, L., Amri, A. & Borden, Katherine L.B. The oncogene eIF4E reprograms the nuclear pore complex to promote mRNA export and oncogenic transformation. Cell Rep 2, 207–215 (2012).
Fung HYJ, Chook YM. Atomic basis of CRM1-cargo recognition, release and inhibition. Semin Cancer Biol. 2014;27:52–61.
Wang AY, Liu H. The past, present, and future of CRM1/XPO1 inhibitors. Stem cell investigation. 2019;6:6–6.
Sun Q, Chen X, Zhou Q, Burstein E, Yang S, Jia D. Inhibiting cancer cell hallmark features through nuclear export inhibition. Signal transduction and targeted therapy. 2016;1:16010.
Sun H, Hattori N, Chien W, Sun Q, Sudo M, E-Ling GL, et al. KPT-330 has antitumour activity against non-small cell lung cancer. Br J Cancer. 2014;111:281–91.
Gandhi UH, Senapedis W, Baloglu E, Unger TJ, Chari A, Vogl D, et al. Clinical implications of targeting XPO1-mediated nuclear export in multiple myeloma. Clin Lymphoma Myeloma Leuk. 2018;18:335–45.
Yang J, Bill MA, Young GS, la Perle K, Landesman Y, Shacham S, et al. Novel small molecule XPO1/CRM1 inhibitors induce nuclear accumulation of TP53, phosphorylated MAPK and apoptosis in human melanoma cells. PLoS One. 2014;9:e102983.
Ranganathan P, Kashyap T, Yu X, Meng X, Lai TH, McNeil B, et al. XPO1 inhibition using selinexor synergizes with chemotherapy in acute myeloid leukemia by targeting DNA repair and restoring topoisomerase IIα to the nucleus. Clin Cancer Res. 2016;22:6142–52.
Garg M, Kanojia D, Mayakonda A, Ganesan TS, Sadhanandhan B, Suresh S, et al. Selinexor (KPT-330) has antitumor activity against anaplastic thyroid carcinoma in vitro and in vivo and enhances sensitivity to doxorubicin. Sci Rep. 2017;7:9749.
clinicaltrials.gov. Studies found for selinexor (2019).
Garzon R, Savona M, Baz R, Andreeff M, Gabrail N, Gutierrez M, et al. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood. 2017;129:3165–74.
Zhang W, et al. Combinatorial targeting of XPO1 and FLT3 exerts synergistic anti-leukemia effects through induction of differentiation and apoptosis in FLT3-mutated acute myeloid leukemias: from concept to clinical trial. Haematologica. 2018;103:1642–53.
Gounder MM, Zer A, Tap WD, Salah S, Dickson MA, Gupta AA, et al. Phase IB study of selinexor, a first-in-class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J Clin Oncol. 2016;34:3166–74.
Vogl DT, Dingli D, Cornell RF, Huff CA, Jagannath S, Bhutani D, et al. Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J Clin Oncol. 2018;36:859–66.
Khoury HJ, Cortes J, Baccarani M, Wetzler M, Masszi T, Digumarti R, et al. Omacetaxine mepesuccinate in patients with advanced chronic myeloid leukemia with resistance or intolerance to tyrosine kinase inhibitors. Leuk Lymphoma. 2015;56:120–7.
Quintas-Cardama A, Kantarjian H, Cortes J. Homoharringtonine, omacetaxine mepesuccinate, and chronic myeloid leukemia circa 2009. Cancer. 2009;115:5382–93.
Tujebajeva RM, Graifer DM, Karpova GG, Ajtkhozhina NA. Alkaloid homoharringtonine inhibits polypeptide chain elongation on human ribosomes on the step of peptide bond formation. FEBS Lett. 1989;257:254–6.
Wetzler M, Segal D. Omacetaxine as an anticancer therapeutic: what is old is new again. Curr Pharm Des. 2011;17:59–64.
Tujebajeva RM, Graifer DM, Matasova NB, Fedorova OS, Odintsov VB, Ajtkhozhina NA, et al. Selective inhibition of the polypeptide chain elongation in eukaryotic cells. Biochim Biophys Acta. 1992;1129:177–82.
Lam SS, et al. Homoharringtonine (omacetaxine mepesuccinate) as an adjunct for FLT3-ITD acute myeloid leukemia. Sci Transl Med. 2016;8:359ra129.
Liu J, Mi Y, Fu M, Yu W, Wang Y, Lin D, et al. Intensive induction chemotherapy with regimen containing intermediate dose cytarabine in the treatment of de novo acute myeloid leukemia. Am J Hematol. 2009;84:422–7.
Jin J, Wang JX, Chen FF, Wu DP, Hu J, Zhou JF, et al. Homoharringtonine-based induction regimens for patients with de-novo acute myeloid leukaemia: a multicentre, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2013;14:599–608.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ipsita Pal, Maryam Safari, Marko Jovanovic, and Susan E. Bates declare that they have no conflict of interest.
Changchun Deng reports grants from TG Therapeutics and Amgen. In addition, Dr. Deng has two patents pending related to the topic reviewed here: 15003-353US0 and IR# CU19337.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on B-cell NHL, T-cell NHL, and Hodgkin Lymphoma
Rights and permissions
About this article

Cite this article
Pal, I., Safari, M., Jovanovic, M. et al. Targeting Translation of mRNA as a Therapeutic Strategy in Cancer. Curr Hematol Malig Rep 14, 219–227 (2019). https://doi.org/10.1007/s11899-019-00530-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-019-00530-y