
Abstract
Purpose of Review
Multiple experimental and clinical studies have identified pathways by which uric acid may facilitate the development and progression of chronic kidney disease (CKD) in people with diabetes. However, it remains uncertain if the association of uric acid with CKD represents a pathogenic effect or merely reflects renal impairment.
Recent Findings
In contrast to many published reports, a recent Mendelian randomization study did not identify a causal link between uric acid and CKD in people with type 1 diabetes. Two recent multicenter randomized control trials, Preventing Early Renal Function Loss in Diabetes (PERL) and FEbuxostat versus placebo rAndomized controlled Trial regarding reduced renal function in patients with Hyperuricemia complicated by chRonic kidney disease stage 3 (FEATHER), were recently designed to assess if uric acid lowering slows progression of CKD.
Summary
We review the evidence supporting a role for uric acid in the pathogenesis of CKD in people with diabetes and the putative benefits of uric acid lowering.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many studies have demonstrated an association between uric acid and kidney disease in patients with diabetes [1,2,3,4,5]. Some investigators have reported a positive correlation between uric acid and the rate of decline in estimated glomerular filtration rate (eGFR), while others have observed only an association with proteinuria. Whether uric acid plays a role in the pathogenesis of chronic kidney disease (CKD) or is simply a marker of metabolic abnormalities remains controversial. This review examines the evidence supporting a role for uric acid in the onset and progression of CKD and the effects of therapeutic lowering of uric acid.
Major Aberrant Metabolic Pathways Leading to Uric Acid Generation in People With Diabetes
In humans, uric acid is the end product of endogenous and exogenous purine metabolism [6•]. Fructose metabolism also generates uric acid [7]. After ingestion, fructose is absorbed into cells and phosphorylated b fructokinase with subsequent depletion of adenosine triphosphate. This leads to increased production of adenosine monophosphate and a rise in serum uric acid [8, 9]. Increases in fructose ingestion may lead to an increase in serum uric acid [10,11,12]. In people with diabetes, uric acid generation from fructose takes on added importance since fructose may be generated endogenously via the polyol pathway [13•].
Why Is Uric Acid Potentially Clinically Relevant?
The serum uric acid level is determined by a balance of uric acid generation, reabsorption, and excretion in the kidney, which is influenced by hydration status and diuretic use [14]. Elevated levels of serum uric acid have been independently associated with cardiovascular disease [15,16,17,18,19], hypertension [18,19,20,21,22], diabetes [23,24,25], metabolic syndrome [24, 26,27,28,29], and CKD [15, 30, 31].
In Vitro Studies: Effects of Uric Acid on Cultured Endothelial Cells
Uric acid impairs basal and vascular endothelial growth factor-induced nitric oxide production in cultured endothelial cells, which reflects induction of inflammatory cascades through the expression of chemokines including monocyte chemoattractant protein-1 and cyclooxygenase-2 [31]. Verzola et al. demonstrated that uric acid increases apoptosis in human proximal tubular cells by triggering a pathway involving nicotinamide adenine dinucleotide phosphate oxidase signaling and urate transporter 1 (URAT1) transport [32].
Effects of Uric Acid on Animal Models
In animal models, uric acid has a plethora of biologic effects including endothelial dysfunction, oxidative stress, vascular smooth muscle cell proliferation, inflammation, activation of phospholipase A2, and increases in TGF-α and TGF-ß1. These changes may lead to tubulointerstitial injury, hypertension, proteinuria, and decreased renal function [31, 33, 34]. Endothelial dysfunction leading to impaired nitric oxide production is mediated, in part, by reactive oxygen species (ROS) [35, 36].
The reaction of xanthine oxidase with xanthine generates superoxide anion and uric acid leading to endothelial dysfunction and hypertension. Khosla et al. demonstrated that induction of mild hyperuricemia by oxonic acid in Sprague Dawley rats induces endothelial dysfunction and a trend toward higher systolic blood pressure by inhibiting nitric oxide production [35]. These investigators also demonstrated that uric acid inhibited nitric oxide production in cultured endothelial cells. The decrease in nitric oxide production induced by uric acid reflects a decrease in serum nitrites and nitrates which is reversed by allopurinol administration [35,36,37]. These data are consistent with hypothesis that hyperuricemia induces endothelial dysfunction.
Since uric acid is a product of xanthine oxidase activity, which generates ROS, it may be a marker of oxidative stress [38]. Therefore, the beneficial effects of allopurinol may be mediated by its active metabolite, oxypurinol, which inhibits production of oxidants by xanthine oxidase. Allopurinol inhibits xanthine oxidase which may reduce formation of ROS and thus have beneficial effects unrelated to serum uric acid levels [14, 38, 39].
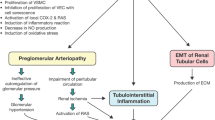
In rat models, hyperuricemia is associated with renal hypertrophy, glomerulosclerosis, interstitial fibrosis, activation of the renin angiotensin system (RAS), and arteriolopathy of the preglomerular renal vasculature [31, 35, 37]. The arteriolopathy may be blunted by administration of allopurinol or the iodine-containing uricosuric agent, benziodarone. Hyperuricemia aggravates kidney injury in diabetes by recruitment of IL-1ß-secreting macrophages, activating nucleotide-binding oligomerization domain protein 3 inflammasomes in macrophages, and promoting chemokine secretion in proximal tubular cells [40]. Hyperuricemia also decreases E-cadherin expression, which may induce an epithelial-to-mesenchymal transition of renal tubular cells [41], the process of differentiation of tubular epithelial cells to myofibroblasts which facilitates the pathogenesis of tubular fibrosis in CKD [42].
Renal Effects of Uric Acid Lowering
Allopurinol administration to mouse models of type 2 diabetes mellitus (KK-Ay/Ta and db/db mice) may blunt proteinuria, tubulointerstitial damage, and decreases in TGF-ß1 [34, 43]. Allopurinol may also partially reverse endothelial dysfunction by reducing intracellular adhesion molecule-1 expression by tubular epithelial cells and blunt hyperuricemia induced changes in E-cadherin and renal fibrosis [43]. Treatment with febuxostat and blocking the metabolism of fructose has been shown to mitigate uric acid-induced pathological changes [44, 45].
Uric Acid and Kidney Disease
Results from cohort studies in the USA, Japan, and Italy provide epidemiologic evidence supporting the hypothesis that uric acid is involved in pathogenesis of kidney disease. Hsu et al. reported results of a cohort study with 5,275,957 person-years of follow-up conducted in patients enrolled in Kaiser Permanente of Northern California. Serum uric acid was an independent risk factor for end-stage renal disease (ESRD) (HR 2.14, 95% CI 1.65–2.77) [30]. Weiner et al. pooled data from 13,338 patients with normal kidney function in two community-based cohorts [46]. Median follow-up was 8.5 ± 0.9 years. Baseline serum uric acid concentrations were higher in the 712 (5.6%) people who developed kidney disease versus those who did not (p < 0.0001). During the study, 712 (5.6%) developed kidney disease defined by an eGFR decrease ≥ 15 mL/min/1.73 m2 with final eGFR < 60 mL/min/1.73 m2, while 302 (2.3%) individuals developed kidney disease defined by creatinine increase (≥ 0.4 mg/dL with final serum creatinine > 1.4 mg/dL in men and 1.2 mg/dL in women). Ohno et al. conducted a 2-year prospective observational study of 2853 healthy Japanese adults aged ≤ 50 years [47]. Changes in serum uric acid levels, even within the normal range, were a sensitive predictor of changes in eGFR (p < 0.001). However, the change in serum uric acid levels (β value of − 0.279) was not sufficient to completely explain the change in eGFR indicating the presence of confounding factors. Moreover, the changes in eGFR and other variables over 2 years were small and could be within expected biologic variation. In a prospective Italian cohort of 900 healthy blood donors followed for 5 years, higher serum uric acid levels were associated with a greater likelihood of eGFR (calculated using the 4-variable Modification of Diet in Renal Disease [MDRD] study equation) decline (HR, 1.13, 95% CI 1.04–1.39) each 1 mg/dL increase in uric acid [48]. Consistent findings have been reported by some but not all studies [49,50,51,52,53,54]. In the MDRD study, the highest tertile of serum uric acid was associated with increased all-cause mortality (HR 1.57, 95% CI 1.07–2.32) but not with kidney failure (HR, 1.20, 95% CI 0.95–1.51) [52]. Sturm et al. reported on a prospective study of 177 Caucasian non-diabetic individuals with CKD and 7 years of follow-up, uric acid was not established as an independent predictor of progression [55]. These discrepant results may reflect differences in study design, definitions of CKD, and criteria for decrease in eGFR.
Uric Acid and Kidney Disease in Patients With Diabetes Mellitus
Renal uric acid handling may be altered in people with diabetes and may differ according to the type of diabetes [13•]. Therefore, we discuss the relationship of uric acid to kidney disease in type 1 and type 2 diabetes individually.
Uric Acid and Kidney Disease in Patients With Type 1 Diabetes Mellitus
Multiple prospective cohort studies in patients with type 1 diabetes mellitus have demonstrated strong associations between baseline uric acid and incidence of albuminuria. Hovind et al. conducted a prospective observational study of 263 patients followed from the onset of type 1 diabetes [2]. Median follow-up was 18 years. The cumulative incidence of persistent micro- and macroalbuminuria was 32.2% (95% CI 25.7–38.7%) and 12.6% (95% CI 7.3–17.9%), respectively. Serum uric acid was independently associated with subsequent development of persistent macroalbuminuria (HR 2.37, 95% CI 1.04–5.37) per 1.68 mg/dL increase in serum uric acid level (p = 0.04), but was not associated with persistent microalbuminuria.
In the Coronary Artery Calcification in Type 1 Diabetes trial (CACTI), a prospective follow-up study, every 1 mg/dL increase in serum uric acid was associated with an independent risk of developing albuminuria (OR 1.8, 95% Cl 1.2–2.8) [3]. Similarly, in the Second Joslin Kidney Study of type 1 diabetes mellitus, serum uric acid levels in the upper portion of the normal range were independently associated with a decreased eGFR [56]. Subsequently, when a subset of patients (n = 355) with albuminuria was followed for 4 to 6 years, there was a dose-response relationship between serum uric acid and risk for an early decrease in eGFR [57].
Mendelian Randomization Studies in Type 1 Diabetes Mellitus
The findings from a recent Mendelian randomization (MR) study cast doubt on the causal relationship of uric acid and progression of kidney disease among patients with type 1 diabetes mellitus while underscoring potential confounding by obesity. The Finnish Diabetic Nephropathy Study group found that baseline serum uric acid was strongly associated with the single nucleotide polymorphisms score but not with any stage of nephropathy based on albuminuria or eGFR. The investigators concluded that there was no evidence of a causal link between serum uric acid and progression of kidney disease [58••]. Whether these results, from a predominantly white population, are generalizable to other ethnicities is uncertain [59]. Furthermore, results from a population-based study of 6184 Caucasians using a bidirectional MR approach suggest that elevated serum uric acid levels may be a consequence of obesity [60]. Another MR study demonstrated that a 1 kg/m2 higher BMI conferred a 28% increased risk in macroalbuminuria, a 43% increased risk of ESRD, and a 33% increased risk of diabetic kidney disease [61]. Therefore, a direct causal link between type 1 diabetes mellitus and uric acid may not exist but rather hyperuricemia may, in part, be a consequence of obesity.
Uric Acid and Kidney Disease in Patients With Type 2 Diabetes Mellitus
Hyperuricemia has been independently associated with incident CKD in patients with type 2 diabetes mellitus [1]. In a 5-year Italian prospective observational study of 1449 participants with normal kidney function at baseline, hyperuricemia was an independent predictor of CKD [5]. In another Italian prospective cohort study of patients with type 2 diabetes mellitus (n = 13,964) with eGFR ≥ 60 mL/min/1.73 m2 and normal urinary albumin excretion rate (UAER), the risk for developing a 10% decrease in eGFR increased linearly across serum uric acid quintiles [1]. Each 1 mg/dL increase in baseline serum uric acid was associated with a 10% increase in risk for developing a decrease in eGFR. In a meta-analysis of nine articles including 20,981 patients with type 2 diabetes, an elevated serum uric acid was associated with an increased risk for kidney disease (OR 1.91, 95% CI 1.07–3.42) [4].
Hyperuricemia may increase progression of CKD. Altemtam et al. conducted a retrospective cohort study of 270 patients with type 2 diabetes and stage 3 and 4 CKD followed at the Sheffield Kidney Institute in the UK from 2000 to 2008 [62]. Mean follow-up was 5.2 ± 1.8 years. The mean rate of decline in eGFR was 1.46 mL/min/1.73 m2/year. Progressive disease was observed in 34.8% patients in whom the mean decline in eGFR was 3.57 ± 1.45 mL/min/1.73 m2/year. Baseline serum uric acid was higher in patients with more rapidly progressive CKD (9.94 ± 1.74 mg/dL) versus those with slower progression (7.63 ± 1.7 mg/dL) (p = 0.004). More recently, Bartakova et al. reported on a prospective cohort of 422 individuals with type 2 diabetes mellitus in the Czech Republic. The median duration of type 2 diabetes was 15 years and median follow-up was 43 months [63]. At baseline, 68% of patients had hyperuricemia, which was associated with an increased rate of CKD progression, independent of the baseline stage of CKD (p < 0.0001).
Kim et al. conducted a retrospective observational longitudinal study of 512 patients with type 2 diabetes in Korea with normal serum uric acid levels and preserved kidney function (eGFR ≥ 60 mL/min/1.73 m2) for a mean follow-up of 3 years. Interestingly, participants with baseline serum uric acid levels in the upper portion of the normal range were at increased risk for developing CKD stage ≥ 3 (HR 2.97, 95% CI 1.15–7.71; p = 0.025) and increased albuminuria (HR 5.52, 95% CI 2.47–12.36; p < 0.001) [64]. Other investigators have reported that high normal serum uric acid levels may be a risk factor for declining renal function in patients with type 2 diabetes mellitus [65, 66].
Serum uric acid may modulate the risk for albuminuria. A Japanese study of 343 men with type 2 diabetes found that serum uric acid concentration was an independent determinant of logarithm of UAER (p < 0.0001) [67]. Similarly, a cross-sectional study of 60 patients in Iran found a positive association of serum uric acid level with proteinuria (p < 0.001) [68]. In addition, a more recent Japanese cohort study of 1802 patients with type 2 diabetes mellitus with normoalbuminuria and eGFR ≥ 60 mL/min/1.73 m2 found that elevated serum uric acid levels were associated with an increased risk for albuminuria but not a decrease in eGFR [69].
Interaction of Insulin and Glucose With Renal Handling of Uric Acid
Renal handling of uric acid is complex. Genome-wide association studies have identified multiple loci, most of which are uric acid transporters [70]. Uric acid is freely filtered across the glomerulus. This is followed by reabsorption and secretion, which coexist along the proximal tubule [70,71,72]. The fractional excretion of uric acid is 8 to 10% [7]. The identification and characterization of these genetically determined uric acid transporters may facilitate the development of novel therapies for lowering serum uric acid.
Insulin facilitates uric acid reabsorption and, not surprisingly, uric acid clearance is inversely correlated with insulin resistance [73]. Administration of sodium glucose cotransporter 2 (SGLT2) inhibitors increase uric acid excretion indirectly enhancing glycosuria, which in turn interacts with GLUT9 isoform 2 along the proximal tubule and possibly in the collecting duct [74].
Multiple factors may explain the association of serum uric acid with eGFR decline and albuminuria in patients with diabetes. Hyperuricemia may be an early manifestation, rather than a risk factor for kidney disease. Obesity is common among patients with diabetes and therefore may be a potential confounder in the observed association of hyperuricemia with kidney disease. Hyperuricemia is a risk factor for hypertension, which in turn increases the risk for kidney disease [22, 75]. Feig et al. suggested that treating hyperuricemia in adolescents with newly diagnosed hypertension was effective at lowering blood pressure [76].
Uric acid may also have direct toxicity increasing the risk for acute kidney injury. Uric acid may induce renal vasoconstriction by inhibiting release of nitric oxide [35, 77]. Although results from animal experiments [35, 37, 78] and observational studies in humans [79] have suggested that uric acid may activate the renin angiotensin system (RAS), a recent randomized controlled trial found that uric acid lowering had no effect on kidney or systemic RAS [80]. Uric acid may also damage the kidney by stimulating an inflammatory response [81, 82]. Urate has both antioxidant and pro-oxidant properties, and although it may be beneficial in scavenging free radicals, it can also impair endothelial function [83].
Effect of Uric Acid Lowering on Kidney Function
Observational studies of the association between uric acid lowering and improved renal outcomes underscore a biologically plausible causative role for uric acid in diabetic kidney disease and identify therapeutic strategies to improve renal outcomes. Multiple studies have assessed the effects of allopurinol on renal disease [84,85,86]. Levy et al. conducted a large, population-based retrospective cohort study (n = 16,186) to assess the effects of serum uric acid lowering on rate of eGFR decline [87]. Patients who achieved serum uric acid < 6 mg/dL had a 37% reduction in outcome events (30% reduction in eGFR or eGFR ≤ 15 mL/min/1.73 m2) (HR 0.63, 95% CI 0.5–0.78; p < 0.0001). Goicoechea et al. conducted a prospective study in which hyperuricemic participants with an eGFR < 60 mL/min/1.73 m2 were randomized to receive allopurinol (100 mg/day) or usual treatment [88]. Mean follow-up was 23.4 ± 7.8 months. At study end, the eGFR had increased minimally in those receiving allopurinol (1.3 ± 1.3 mL/min/1.73 m2; NS) but decreased in the control arm (3.3 ± 1.2 mL/min/1.73 m2; p = 0.018). Allopurinol also reduced the risk for cardiovascular events and all-cause hospitalization.
Similar results were observed in an open parallel study in China where 176 participants with type 2 diabetes were randomized to allopurinol or standard treatment [89••]. After 3 years, the decline in eGFR in the allopurinol group was 0.8 ± 3.9 mL/min/1.73 m2 versus 4.9 ± 5 mL/min/1.73 m2 in the conventional group (p < 0.001). The decrease in albuminuria was greater in the allopurinol group (p < 0.01).
Results of a meta-analysis of eight randomized controlled trials (RCTs) with a total of 476 participants suggested that the effects of allopurinol on proteinuria, eGFR, and progression to ESRD are inconclusive [90]. In five trials, there were no significant differences in the changes in eGFR between the allopurinol and control arms (mean difference 3.1 mL/min/1.73 m2; p = 0.1). In contrast, in three trials, the changes in serum creatinine from baseline favored allopurinol (mean difference 0.4 mg/dL; p = 0.03). In another meta-analysis of 19 RCTs with 992 participants, the pooled estimate of eGFR (mean difference 3.2 mL/min/1.73 m2; p = 0.04) favored allopurinol, while the changes in proteinuria were similar across arms (p = 0.58) [39]. In an Iranian RCT with 40 participants with type 2 diabetes, low-dose allopurinol treatment for 4 months reduced proteinuria from 1756 mg/24 h to 1011 mg/24 h (p = 0.049) [91].
Recent Studies That Assessed the Effects of Serum Uric Acid Lowering
Two recent multicenter RCTs were designed to assess the renal protection afforded by uric acid lowering. The Preventing Early Renal Function Loss in Diabetes (PERL) trial is an ongoing multicenter, double-blind, placebo-controlled RCT that is comparing the effects of allopurinol versus placebo on decline in eGFR to determine if uric acid lowering slows progression of CKD in participants with type 1 diabetes mellitus (ClinicalTrials.gov Identifier NCT02017171) [92, 93]. Eligibility criteria are an increased UAER (30–5000 mg/24 h if not on RAS blocking agent or 18–5000 mg/24 h if on RAS blocking agent) or a decreasing eGFR (≥ 3 mL/min/1.73m2/year). Estimated GFR was chosen as the primary outcome since it may decline in the absence of microalbuminuria [94, 95]. The majority of the patients with microalbuminuria may regress to normoalbuminuria but early eGFR loss usually progresses to CKD and ESRD [95, 96]. The study is important because there is clinical equipoise and the risk of significant adverse reactions is low [97].
The FEATHER trial (FEbuxostat versus placebo rAndomized controlled Trial regarding reduced renal function in patients with Hyperuricemia complicated by chRonic kidney disease stage 3) is being conducted in asymptomatic hyperuricemic Japanese patients with CKD stage 3 (UMIN Clinical Trials Registry, Unique trial number UMIN000008343). It was designed to assess the effects of febuxostat in moderately controlled diabetes (HgbA1C < 8%) and participants without diabetes [98, 99]. In comparison to allopurinol, febuxostat provides more selective and potent inhibition of xanthine oxidase with greater hypouricemic activity [100]. Febuxostat is excreted primarily through the liver, whereas allopurinol is excreted by the kidney. The Cardiovascular Safety of Febuxostat and Allopurinol in Patients with Gout and Cardiovascular Comorbidities (CARES) trial is the first randomized, controlled clinical trial that evaluated the long-term safety of xanthine oxidase inhibitors in gout patients with a history of prior CV events or disease (ClinicalTrials.gov Identifier NCT01101035). Although the trial has been completed, the results have not yet been published. However, the United States Food and Drug Administration has issued a preliminary warning of increased risk of cardiovascular and all-cause mortality with the use of febuxostat compared to allopurinol [101,101,••, 102, 103].
A post hoc analysis of the Febuxostat Open-Label Clinical Trial of Urate-Lowering Efficacy and Safety Study (FOCUS) with 116 hyperuricemic patients treated with febuxostat for 5 years found an inverse correlation between uric acid and eGFR and projected an improvement in eGFR of 1 mL/min/1.73 m2 for every 1 mg/dL decrease in serum uric acid [104]. In a placebo controlled RCT, conducted in Eastern India, patients with stage 3 and 4 CKD and asymptomatic hyperuricemia were randomized to febuxostat (n = 45) versus placebo (n = 48) for 6 months [105]. Estimated GFR decreased in the placebo group (32.6 ± 11.6 to 28.2 ± 11.5 mL/min/1.73 m2; p = 0.003) but not in the febuxostat group (31.5 ± 13.6 to 33.7 ± 16.6 mL/min/1.73 m2; NS). The percentage of participants who experienced ≥ 10% decrease was greater in the placebo (54%) versus the febuxostat group (38%).
SGLT2 inhibitors have been shown to lower uric acid levels. Zinman et al. conducted a RCT using the SGLT2 inhibitor empagliflozin or placebo in 7020 patients for a median duration of 3.1 years and found lower rates of death from cardiovascular causes, hospitalization from any cause and all-cause mortality. It was also associated with reduction in diastolic and systolic blood pressure, hemoglobin A1C, and serum uric acid levels [106]. In a meta-analysis of 62 studies consisting of 34,951 participants, SGLT2 inhibitors decreased serum uric acid by a range of 17.4 mmol/L (0.29 mg/dL) to 45.8 mmol/L (0.77 mg/dL) with a weighted mean difference of 37.73 mmol/L (0.63 mg/dL). However, the decline in serum uric acid was blunted in patients with longer duration of diabetes, higher hemoglobin A1C, and patients with eGFR < 60 mL/min/1.73m2. These small reductions in serum uric acid are unlikely to explain the cardiovascular benefits seen with the use of these drugs [107].
A post hoc analysis of 1342 participants with type 2 diabetes and nephropathy who participated in the Reduction of End Points in Non-Insulin-Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan (RENAAL) trial (1342 participants with diabetic nephropathy, median follow-up 3.4 years) found that the risk of adverse renal outcomes was decreased by 6% for every 0.5 mg/dL decrease in serum uric acid levels during the first 6 months of treatment with losartan [108]. This effect accounted for about 20% of the renoprotective effect of losartan.
Conclusion
There is substantial evidence from observational studies that supports a significant association between higher serum uric acid levels and the risk for CKD among people with diabetes mellitus. Surprisingly however, the results of clinical trials to date have not conclusively demonstrated a beneficial effect of serum uric acid lowering. Despite the current availability of conclusive trial data, Barkatoba et al. have recommended 10 to 15% lower serum uric acid cutoff values for people with diabetes mellitus (6.35 mg/dL for men and 5.2 mg/dL for women) to confer protection against kidney disease [63]. Results from the PERL and FEATHER trials may better define the potential benefit of lowering serum uric acid [93, 98, 99].
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
De CS, Viazzi F, Pacilli A, Giorda C, Ceriello A, Gentile S, et al. Serum uric acid and risk of CKD in type 2 diabetes. Clin J Am Soc Nephrol. 2015;10:1921–9.
Hovind P, Rossing P, Tarnow L, Johnson RJ, Parving HH. Serum uric acid as a predictor for development of diabetic nephropathy in type 1 diabetes: an inception cohort study. Diabetes. 2009;58:1668–71.
Jalal DI, Rivard CJ, Johnson RJ, Maahs DM, McFann K, Rewers M, et al. Serum uric acid levels predict the development of albuminuria over 6 years in patients with type 1 diabetes: findings from the Coronary Artery Calcification in Type 1 Diabetes study. Nephrol Dial Transplant. 2010;25:1865–9.
Xu Y, Zhu J, Gao L, Liu Y, Shen J, Shen C, et al. Hyperuricemia as an independent predictor of vascular complications and mortality in type 2 diabetes patients: a meta-analysis. PLoS One. 2013;8:e78206.
Zoppini G, Targher G, Chonchol M, Ortalda V, Abaterusso C, Pichiri I, et al. Serum uric acid levels and incident chronic kidney disease in patients with type 2 diabetes and preserved kidney function. Diabetes Care. 2012;35:99–104.
• Maiuolo J, Oppedisano F, Gratteri S, Muscoli C, Mollace V. Regulation of uric acid metabolism and excretion. Int J Cardiol. 2016;213:8–14. Reviews the several enzymatic pathways for uric acid production.
Mende C. Management of chronic kidney disease: the relationship between serum uric acid and development of nephropathy. Adv Ther. 2015;32:1177–91.
Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M, et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes. 2013;62:3307–15.
Lytvyn Y, Perkins BA, Cherney DZ. Uric acid as a biomarker and a therapeutic target in diabetes. Can J Diabetes. 2015;39:239–46.
Choi JW, Ford ES, Gao X, Choi HK. Sugar-sweetened soft drinks, diet soft drinks, and serum uric acid level: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2008;59:109–16.
Cox CL, Stanhope KL, Schwarz JM, Graham JL, Hatcher B, Griffen SC, et al. Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr Metab (Lond). 2012;9:68.
Stirpe F, Della CE, Bonetti E, Abbondanza A, Abbati A, De SF. Fructose-induced hyperuricaemia. Lancet. 1970;2:1310–1.
• Bjornstad P, Lanaspa MA, Ishimoto T, Kosugi T, Kume S, Jalal D, et al. Fructose and uric acid in diabetic nephropathy. Diabetologia. 2015;58:1993–2002. Discusses the relationship between uric acid and fructose in the development of diabetic nephropathy.
Kang DH, Chen W. Uric acid and chronic kidney disease: new understanding of an old problem. Semin Nephrol. 2011;31:447–52.
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2008;359:1811–21.
Kanbay M, Segal M, Afsar B, Kang DH, Rodriguez-Iturbe B, Johnson RJ. The role of uric acid in the pathogenesis of human cardiovascular disease. Heart. 2013;99:759–66.
Liu WC, Hung CC, Chen SC, Yeh SM, Lin MY, Chiu YW, et al. Association of hyperuricemia with renal outcomes, cardiovascular disease, and mortality. Clin J Am Soc Nephrol. 2012;7:541–8.
Kuwabara M. Hyperuricemia, cardiovascular disease, and hypertension. Pulse (Basel). 2016;3:242–52.
Tangri N, Weiner DE. Uric acid, CKD, and cardiovascular disease: confounders, culprits, and circles. Am J Kidney Dis. 2010;56:247–50.
Masuo K, Kawaguchi H, Mikami H, Ogihara T, Tuck ML. Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension. 2003;42:474–80.
Mazzali M, Kanbay M, Segal MS, Shafiu M, Jalal D, Feig DI, et al. Uric acid and hypertension: cause or effect? Curr Rheumatol Rep. 2010;12:108–17.
Mellen PB, Bleyer AJ, Erlinger TP, Evans GW, Nieto FJ, Wagenknecht LE, et al. Serum uric acid predicts incident hypertension in a biethnic cohort: the atherosclerosis risk in communities study. Hypertension. 2006;48:1037–42.
Dehghan A, van HM, Sijbrands EJ, Hofman A, Witteman JC. High serum uric acid as a novel risk factor for type 2 diabetes. Diabetes Care. 2008;31:361–2.
Li C, Hsieh MC, Chang SJ. Metabolic syndrome, diabetes, and hyperuricemia. Curr Opin Rheumatol. 2013;25:210–6.
Shani M, Vinker S, Dinour D, Leiba M, Twig G, Holtzman EJ, et al. High normal uric acid levels are associated with an increased risk of diabetes in lean, normoglycemic healthy women. J Clin Endocrinol Metab. 2016;101:3772–8.
Krishnan E, Pandya BJ, Chung L, Hariri A, Dabbous O. Hyperuricemia in young adults and risk of insulin resistance, prediabetes, and diabetes: a 15-year follow-up study. Am J Epidemiol. 2012;176:108–16.
Lai HM, Chen CJ, Su BY, Chen YC, Yu SF, Yen JH, et al. Gout and type 2 diabetes have a mutual inter-dependent effect on genetic risk factors and higher incidences. Rheumatology (Oxford). 2012;51:715–20.
Sui X, Church TS, Meriwether RA, Lobelo F, Blair SN. Uric acid and the development of metabolic syndrome in women and men. Metabolism. 2008;57:845–52.
Yu TY, Jee JH, Bae JC, Jin SM, Baek JH, Lee MK, et al. Serum uric acid: a strong and independent predictor of metabolic syndrome after adjusting for body composition. Metabolism. 2016;65:432–40.
Hsu CY, Iribarren C, McCulloch CE, Darbinian J, Go AS. Risk factors for end-stage renal disease: 25-year follow-up. Arch Intern Med. 2009;169:342–50.
Kang DH, Nakagawa T, Feng L, Watanabe S, Han L, Mazzali M, et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol. 2002;13:2888–97.
Verzola D, Ratto E, Villaggio B, Parodi EL, Pontremoli R, Garibotto G, et al. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS One. 2014;9:e115210.
Diwan V, Mistry A, Gobe G, Brown L. Adenine-induced chronic kidney and cardiovascular damage in rats. J Pharmacol Toxicol Methods. 2013;68:197–207.
Kim SM, Choi YW, Seok HY, Jeong KH, Lee SH, Lee TW, et al. Reducing serum uric acid attenuates TGF-beta1-induced profibrogenic progression in type 2 diabetic nephropathy. Nephron Exp Nephrol. 2012;121:e109–21.
Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005;67:1739–42.
Puddu P, Puddu GM, Cravero E, Vizioli L, Muscari A. Relationships among hyperuricemia, endothelial dysfunction and cardiovascular disease: molecular mechanisms and clinical implications. J Cardiol. 2012;59:235–42.
Mazzali M, Kanellis J, Han L, Feng L, Xia YY, Chen Q, et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Ren Physiol. 2002;282:F991–7.
Jalal DI, Maahs DM, Hovind P, Nakagawa T. Uric acid as a mediator of diabetic nephropathy. Semin Nephrol. 2011;31:459–65.
Kanji T, Gandhi M, Clase CM, Yang R. Urate lowering therapy to improve renal outcomes in patients with chronic kidney disease: systematic review and meta-analysis. BMC Nephrol. 2015;16:58.
Kim SM, Lee SH, Kim YG, Kim SY, Seo JW, Choi YW, et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am J Physiol Ren Physiol. 2015;308:F993–F1003.
Ryu ES, Kim MJ, Shin HS, Jang YH, Choi HS, Jo I, et al. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am J Physiol Ren Physiol. 2013;304:F471–80.
Yang J, Liu Y. Blockage of tubular epithelial to myofibroblast transition by hepatocyte growth factor prevents renal interstitial fibrosis. J Am Soc Nephrol. 2002;13:96–107.
Kosugi T, Nakayama T, Heinig M, Zhang L, Yuzawa Y, Sanchez-Lozada LG, et al. Effect of lowering uric acid on renal disease in the type 2 diabetic db/db mice. Am J Physiol Ren Physiol. 2009;297:F481–8.
Lanaspa MA, Ishimoto T, Cicerchi C, Tamura Y, Roncal-Jimenez CA, Chen W, et al. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J Am Soc Nephrol. 2014;25:2526–38.
Sanchez-Lozada LG, Tapia E, Soto V, Avila-Casado C, Franco M, Wessale JL, et al. Effect of febuxostat on the progression of renal disease in 5/6 nephrectomy rats with and without hyperuricemia. Nephron Physiol. 2008;108:69–78.
Weiner DE, Tighiouart H, Elsayed EF, Griffith JL, Salem DN, Levey AS. Uric acid and incident kidney disease in the community. J Am Soc Nephrol. 2008;19:1204–11.
Ohno M, Deguchi F, Izumi K, Ishigaki H, Sarui H, Sasaki A, et al. Correlation between renal function and common risk factors for chronic kidney disease in a healthy middle-aged population: a prospective observational 2-year study. PLoS One. 2014;9:e113263.
Bellomo G, Venanzi S, Verdura C, Saronio P, Esposito A, Timio M. Association of uric acid with change in kidney function in healthy normotensive individuals. Am J Kidney Dis. 2010;56:264–72.
Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res. 2001;24:691–7.
Iseki K, Ikemiya Y, Inoue T, Iseki C, Kinjo K, Takishita S. Significance of hyperuricemia as a risk factor for developing ESRD in a screened cohort. Am J Kidney Dis. 2004;44:642–50.
Langford HG, Blaufox MD, Borhani NO, Curb JD, Molteni A, Schneider KA, et al. Is thiazide-produced uric acid elevation harmful? Analysis of data from the hypertension detection and follow-up program. Arch Intern Med. 1987;147:645–9.
Madero M, Sarnak MJ, Wang X, Greene T, Beck GJ, Kusek JW, et al. Uric acid and long-term outcomes in CKD. Am J Kidney Dis. 2009;53:796–803.
Obermayr RP, Temml C, Gutjahr G, Knechtelsdorfer M, Oberbauer R, Klauser-Braun R. Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol. 2008;19:2407–13.
Sonoda H, Takase H, Dohi Y, Kimura G. Uric acid levels predict future development of chronic kidney disease. Am J Nephrol. 2011;33:352–7.
Sturm G, Kollerits B, Neyer U, Ritz E, Kronenberg F. Uric acid as a risk factor for progression of non-diabetic chronic kidney disease? The Mild to Moderate Kidney Disease (MMKD) Study. Exp Gerontol. 2008;43:347–52.
Rosolowsky ET, Ficociello LH, Maselli NJ, Niewczas MA, Binns AL, Roshan B, et al. High-normal serum uric acid is associated with impaired glomerular filtration rate in nonproteinuric patients with type 1 diabetes. Clin J Am Soc Nephrol. 2008;3:706–13.
Ficociello LH, Rosolowsky ET, Niewczas MA, Maselli NJ, Weinberg JM, Aschengrau A, et al. High-normal serum uric acid increases risk of early progressive renal function loss in type 1 diabetes: results of a 6-year follow-up. Diabetes Care. 2010;33:1337–43.
•• Ahola AJ, Sandholm N, Forsblom C, Harjutsalo V, Dahlstrom E, Groop PH. The serum uric acid concentration is not causally linked to diabetic nephropathy in type 1 diabetes. Kidney Int. 2017;91:1178–85. Mendelian randomization study that concluded that there was no causal link between serum uric acid and progression of kidney disease in type 1 diabetes.
Gul A, Harford A, Zager P. Mendelian randomization to establish the causality of uric acid with diabetic nephropathy in type 1 diabetics. Kidney Int. 2017;91:1005–7.
Lyngdoh T, Vuistiner P, Marques-Vidal P, Rousson V, Waeber G, Vollenweider P, et al. Serum uric acid and adiposity: deciphering causality using a bidirectional Mendelian randomization approach. PLoS One. 2012;7:e39321.
Todd JN, Dahlstrom EH, Salem RM, Sandholm N, Forsblom C, McKnight AJ, et al. Genetic evidence for a causal role of obesity in diabetic kidney disease. Diabetes. 2015;64:4238–46.
Altemtam N, Russell J, El NM. A study of the natural history of diabetic kidney disease (DKD). Nephrol Dial Transplant. 2012;27:1847–54.
Bartakova V, Kuricova K, Pacal L, Nova Z, Dvorakova V, Svrckova M, et al. Hyperuricemia contributes to the faster progression of diabetic kidney disease in type 2 diabetes mellitus. J Diabetes Complicat. 2016;30:1300–7.
Kim WJ, Kim SS, Bae MJ, Yi YS, Jeon YK, Kim BH, et al. High-normal serum uric acid predicts the development of chronic kidney disease in patients with type 2 diabetes mellitus and preserved kidney function. J Diabetes Complicat. 2014;28:130–4.
Chang YH, Lei CC, Lin KC, Chang DM, Hsieh CH, Lee YJ. Serum uric acid level as an indicator for CKD regression and progression in patients with type 2 diabetes mellitus—a 4.6-year cohort study. Diabetes Metab Res Rev. 2016;32:557–64.
Tanaka K, Hara S, Hattori M, Sakai K, Onishi Y, Yoshida Y, et al. Role of elevated serum uric acid levels at the onset of overt nephropathy in the risk for renal function decline in patients with type 2 diabetes. J Diabetes Investig. 2015;6:98–104.
Fukui M, Tanaka M, Shiraishi E, Harusato I, Hosoda H, Asano M, et al. Serum uric acid is associated with microalbuminuria and subclinical atherosclerosis in men with type 2 diabetes mellitus. Metabolism. 2008;57:625–9.
Behradmanesh S, Horestani MK, Baradaran A, Nasri H. Association of serum uric acid with proteinuria in type 2 diabetic patients. J Res Med Sci. 2013;18:44–6.
Takagi M, Babazono T, Uchigata Y. Differences in risk factors for the onset of albuminuria and decrease in glomerular filtration rate in people with type 2 diabetes mellitus: implications for the pathogenesis of diabetic kidney disease. Diabet Med. 2015;32:1354–60.
Anzai N, Endou H. Urate transporters: an evolving field. Semin Nephrol. 2011;31:400–9.
Bobulescu IA, Moe OW. Renal transport of uric acid: evolving concepts and uncertainties. Adv Chronic Kidney Dis. 2012;19:358–71.
Reginato AM, Mount DB, Yang I, Choi HK. The genetics of hyperuricaemia and gout. Nat Rev Rheumatol. 2012;8:610–21.
Perez-Ruiz F, Aniel-Quiroga MA, Herrero-Beites AM, Chinchilla SP, Erauskin GG, Merriman T. Renal clearance of uric acid is linked to insulin resistance and lower excretion of sodium in gout patients. Rheumatol Int. 2015;35:1519–24.
Chino Y, Samukawa Y, Sakai S, Nakai Y, Yamaguchi J, Nakanishi T, et al. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm Drug Dispos. 2014;35:391–404.
Krishnan E, Kwoh CK, Schumacher HR, Kuller L. Hyperuricemia and incidence of hypertension among men without metabolic syndrome. Hypertension. 2007;49:298–303.
Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA. 2008;300:924–32.
Kang DH, Park SK, Lee IK, Johnson RJ. Uric acid-induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol. 2005;16:3553–62.
Sanchez-Lozada LG, Tapia E, Lopez-Molina R, Nepomuceno T, Soto V, Avila-Casado C, et al. Effects of acute and chronic L-arginine treatment in experimental hyperuricemia. Am J Physiol Ren Physiol. 2007;292:F1238–44.
Perlstein TS, Gumieniak O, Hopkins PN, Murphey LJ, Brown NJ, Williams GH, et al. Uric acid and the state of the intrarenal renin-angiotensin system in humans. Kidney Int. 2004;66:1465–70.
McMullan CJ, Borgi L, Fisher N, Curhan G, Forman J. Effect of uric acid lowering on renin-angiotensin-system activation and ambulatory BP: a randomized controlled trial. Clin J Am Soc Nephrol. 2017;12:807–16.
Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:2199–210.
Friedewald JJ, Rabb H. Inflammatory cells in ischemic acute renal failure. Kidney Int. 2004;66:486–91.
Krishnan E. Inflammation, oxidative stress and lipids: the risk triad for atherosclerosis in gout. Rheumatology (Oxford). 2010;49:1229–38.
Kanbay M, Ozkara A, Selcoki Y, Isik B, Turgut F, Bavbek N, et al. Effect of treatment of hyperuricemia with allopurinol on blood pressure, creatinine clearence, and proteinuria in patients with normal renal functions. Int Urol Nephrol. 2007;39:1227–33.
Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47:51–9.
Talaat KM, el-Sheikh AR. The effect of mild hyperuricemia on urinary transforming growth factor beta and the progression of chronic kidney disease. Am J Nephrol. 2007;27:435–40.
Levy GD, Rashid N, Niu F, Cheetham TC. Effect of urate-lowering therapies on renal disease progression in patients with hyperuricemia. J Rheumatol. 2014;41:955–62.
Goicoechea M, de Vinuesa SG, Verdalles U, Ruiz-Caro C, Ampuero J, Rincon A, et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010;5:1388–93.
•• Liu P, Chen Y, Wang B, Zhang F, Wang D, Wang Y. Allopurinol treatment improves renal function in patients with type 2 diabetes and asymptomatic hyperuricemia: 3-year randomized parallel-controlled study. Clin Endocrinol (Oxf). 2015;83:475–82. Randomized controlled trial in patients with type 2 diabetes which showed decrease in urinary albumin excretion rate and renal protective effects in the allopurinol group.
Bose B, Badve SV, Hiremath SS, Boudville N, Brown FG, Cass A, et al. Effects of uric acid-lowering therapy on renal outcomes: a systematic review and meta-analysis. Nephrol Dial Transplant. 2014;29:406–13.
Momeni A, Shahidi S, Seirafian S, Taheri S, Kheiri S. Effect of allopurinol in decreasing proteinuria in type 2 diabetic patients. Iran J Kidney Dis. 2010;4:128–32.
Online Source: National Institutes of Health, United States. A multicenter clinical trial of allopurinol to prevent kidney function loss in type 1 diabetics. ClinicalTrials.gov Identifier NTC02017171; https://clinicaltrials.gov/ct2/show/NCT02017171. last updated 03/07/2017. Accessed 4 Dec 2017.
Maahs DM, Caramori L, Cherney DZ, Galecki AT, Gao C, Jalal D, et al. Uric acid lowering to prevent kidney function loss in diabetes: the preventing early renal function loss (PERL) allopurinol study. Curr Diab Rep. 2013;13:550–9.
Molitch ME, Steffes M, Sun W, Rutledge B, Cleary P, de Boer IH, et al. Development and progression of renal insufficiency with and without albuminuria in adults with type 1 diabetes in the diabetes control and complications trial and the epidemiology of diabetes interventions and complications study. Diabetes Care. 2010;33:1536–43.
Perkins BA, Ficociello LH, Ostrander BE, Silva KH, Weinberg J, Warram JH, et al. Microalbuminuria and the risk for early progressive renal function decline in type 1 diabetes. J Am Soc Nephrol. 2007;18:1353–61.
Caramori ML, Fioretto P, Mauer M. The need for early predictors of diabetic nephropathy risk: is albumin excretion rate sufficient? Diabetes. 2000;49:1399–408.
Paisansinsup T, Breitenstein MK, Schousboe JT. Association between adverse reactions to allopurinol and exposures to high maintenance doses: implications for management of patients using allopurinol. J Clin Rheumatol. 2013;19:180–6.
Online Source: UMIN Clinical Trials Registry, Japan. FEbuxostat versus placebo rAndomized controlled Trial (FEATHER study). Unique trial number UMIN000008343; http://www.umin.ac.jp/ctr/index.htm. last updated 12/17/2015. Accessed 14 Sept2017.
Hosoya T, Kimura K, Itoh S, Inaba M, Uchida S, Tomino Y, et al. The effect of febuxostat to prevent a further reduction in renal function of patients with hyperuricemia who have never had gout and are complicated by chronic kidney disease stage 3: study protocol for a multicenter randomized controlled study. Trials. 2014;15:26.
Ye P, Yang S, Zhang W, Lv Q, Cheng Q, Mei M, et al. Efficacy and tolerability of febuxostat in hyperuricemic patients with or without gout: a systematic review and meta-analysis. Clin Ther. 2013;35:180–9.
Online Source: Department of Health and Human Services, United States. FDA to evaluate increased risk of heart-related death and death from all causes with the gout medicine febuxostat (Uloric). FDA Drug Safety Communication; https://www.fda.gov/Drugs/DrugSafety/ucm584702.htm. last updated 11/15/2017. Accessed 20 Nov 2017. The United States Food and Drug Administration warning regarding increased risk of cardiovascular and all-cause mortality in patients using febuxostat as compared to allopurinol.
Online Source: National Institutes of Health, United States. Cardiovascular safety of febuxostat and allopurinol in patients with gout and cardiovascular comorbidities (CARES). ClinicalTrials.gov Identifier NCT01101035; https://clinicaltrials.gov/ct2/show/NCT01101035. last updated 08/11/2017. Accessed 20 Nov2017.
White WB, Chohan S, Dabholkar A, Hunt B, Jackson R. Cardiovascular safety of febuxostat and allopurinol in patients with gout and cardiovascular comorbidities. Am Heart J. 2012;164:14–20.
Whelton A, MacDonald PA, Zhao L, Hunt B, Gunawardhana L. Renal function in gout: long-term treatment effects of febuxostat. J Clin Rheumatol. 2011;17:7–13.
Sircar D, Chatterjee S, Waikhom R, Golay V, Raychaudhury A, Chatterjee S, et al. Efficacy of febuxostat for slowing the GFR decline in patients with CKD and asymptomatic hyperuricemia: a 6-month, double-blind, randomized, placebo-controlled trial. Am J Kidney Dis. 2015;66:945–50.
Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–28.
van Bommel EJ, Muskiet MH, Tonneijck L, Kramer MH, Nieuwdorp M, van Raalte DH. SGLT2 inhibition in the diabetic kidney—from mechanisms to clinical outcome. Clin J Am Soc Nephrol. 2017;12:700–10.
Miao Y, Ottenbros SA, Laverman GD, Brenner BM, Cooper ME, Parving HH, et al. Effect of a reduction in uric acid on renal outcomes during losartan treatment: a post hoc analysis of the reduction of endpoints in non-insulin-dependent diabetes mellitus with the Angiotensin II Antagonist Losartan Trial. Hypertension. 2011;58:2–7.
Acknowledgments
We would like to thank Ms. Leslie Firkins and Ms. Serena Cumber for their help in preparation of this manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Dialysis Clinic, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ambreen Gul is an employee of Dialysis Clinic, Inc.
Philip Zager is an employee of the University of New Mexico and Dialysis Clinic, Inc.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Microvascular Complications—Nephropathy
Rights and permissions
About this article

Cite this article
Gul, A., Zager, P. Does Altered Uric Acid Metabolism Contribute to Diabetic Kidney Disease Pathophysiology?. Curr Diab Rep 18, 18 (2018). https://doi.org/10.1007/s11892-018-0985-5
Published:
DOI: https://doi.org/10.1007/s11892-018-0985-5