
Abstract
The prevalence of type 2 diabetes is increasing worldwide, and while numerous treatments exist, none of the current pharmacologic therapies is curative. Pharmacologic approaches that increase beta cell mass may present an avenue for actual cure. There have been numerous reports on factors that can induce beta cell proliferation in rodents, whereas there are still very limited data on the occurrence of beta cell proliferation in humans. The recent discovery of the hormone betatrophin, which in mice counteracted glucose intolerance induced by insulin resistance by potently stimulating beta cell proliferation, has boosted the hope for a new target for drug development for the treatment of diabetes mellitus in humans. With the encouraging preclinical findings as a background, this review presents the available clinical data on betatrophin and discusses its possible role in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In 2011, the global prevalence of diabetes was estimated by the International Diabetes Federation to be 366 million, and the number is estimated to reach 552 million by 2030 [1]. A common denominator for both type 1 and type 2 diabetes is the loss of functional beta cell mass. In humans, a curvilinear relationship between beta cell mass and fasting blood glucose concentration has been shown [2]. Autopsy studies of patients with type 2 diabetes have demonstrated a mean beta cell deficit of 40–60 % [3, 4], but there are also reports on unaltered anatomical beta cell mass in type 2 diabetes [5, 6]. At onset of type 1 diabetes, beta cell mass has regularly decreased to 20–40 % of normal [7], and the number of beta cells continues to decrease. However, worthy to note is that small numbers of insulin-producing beta cells persist and function in a significant percentage of patients with type 1 diabetes even decades after onset of disease, which suggests either cessation of immune attack or the existence of a pool of beta cells resistant to destruction [8–10]. In both type 1 and type 2 diabetes research, the means to obtain human beta cell proliferation are therefore of enormous interest. A hormone or drug that could be administered in order to induce beta cell proliferation and thereby restore beta cell mass would be a potential cure for both type 1 and type 2 diabetes. There have been numerous reports on hormones and drugs that can induce beta cell proliferation in mice and rats, whereas there are still very limited data on the occurrence of beta cell proliferation in humans.
Beta Cell Proliferation
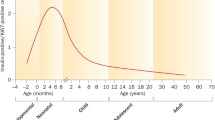
Studies of rats and mice have provided many insights into the development and physiology of the mammalian beta cell, but with regard to proliferation, marked differences exist between species [11–13]. In humans, the peak of beta cell proliferation at any point during life is approximately 2 %, whereas in rodents, this peak is considerably higher, with neonatal beta cell replication in the range of 20 % [14]. Proliferation of human beta cells begins shortly after birth, continues at its highest rate during the first year, and then rapidly declines already during early childhood, reaching a proliferation index that approaches zero in adulthood [15–17]. Tested stimuli known to remarkably induce beta cell proliferation in adult rodents such as different growth factors, laminins, partial pancreatectomy, pregnancy, and high-fat feeding have proven ineffective to induce proliferation of adult human beta cells [18–21]. In fact, merely a few factors such as glucose [22], γ-aminobutyric acid [23], and signals from inflammatory cells [24, 25] or neural crest stem cells [26] have this far been reported to have some human beta cell mitogenic effects. Only by the use of gene therapy techniques, where cdk6 and cyclin D1 were overexpressed, it has in vitro been shown that adult human beta cells can be induced to strongly proliferate, and in a regulated manner without de-differentiation of cells [27–29]. Although being proof-of-principle studies, gene therapy techniques are unlikely to be directly acceptable for human diabetes therapy, and other approaches are clearly needed.
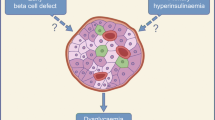
Cumulative research has demonstrated that beta cells in rodents require a connection with surrounding cells and organs for development, for proliferation, and to maintain function and mass homeostasis [30, 31]. Therefore, the description of the hormone betatrophin in mice, produced predominantly in liver and fat tissues in insulin-resistant conditions and acting as a very potent stimulator of beta cell proliferation [32••], has recently attracted considerable interest. Such signal would be a logic feedback to the pancreatic islets during insulin-resistant conditions causing a compensatory increase in beta cell mass. Indeed, an increased beta cell mass has been observed in autopsy pancreas specimens from obese humans, although no concomitant beta cell proliferation has been observed [33]. The latter finding may, however, be erroneous due to a postmortem decline in replication markers such as Ki67 [34]. There are studies showing that adult human beta cells may proliferate in response to an obesogenic environment when transplanted in mice [35, 36].
Betatrophin
The hormone betatrophin was initially described under the name hepatocellular carcinoma-associated protein TD26 [37]. It has also been described under the names lipasin [38], refeeding-induced fat and liver (RIFL) [39], and angiopoietin-like protein 8 [40]. In this review, we will, however, refer to the hormone as betatrophin, a name given by Yi et al. due to its stimulatory effect on beta cell proliferation [32••]. The hormone belongs to the angiopoietin-like protein family and is encoded by the C19orf80 gene (in a mouse named Gm6484). In mouse, it is predominantly expressed in the liver, white adipose tissue, and brown adipose tissue, whereas in human, it is expressed mainly in the liver only [38–40]. The expression of betatrophin in mice is regulated by both nutrition (increased expression in response to a high-fat diet) and temperature (increased expression in a cold environment) [41]. Yi et al. treated the mice with an insulin receptor antagonist (S961) for 1 week to induce glucose intolerance and hyperinsulinemia [32••]. Concomitantly, there was a dose-dependent increase in beta cell proliferation, as evaluated by Ki67 immunohistochemical staining. Four days after the S961 treatment was stopped, proliferation rates returned to baseline levels. No effect on proliferation was observed when isolated islets from the mice were incubated with S961. Microarray analysis of the liver, white adipose tissue, skeletal muscle, and pancreatic islets identified the gene encoding betatrophin in the mouse (Gm6484), and by real-time PCR, the mRNA expression was found to be increased in liver and white adipose tissue in the animals treated with S961. The mRNA expression of betatrophin was also examined in ob/ob mice, db/db mice, and in pregnant mice and was found to be increased in all of these conditions. However, no increase in betatrophin was observed in mice with acute loss of beta cells due to beta cell specific diphtheria toxin expression. The authors also showed that betatrophin is a secreted hormone that can be detected in human plasma. Finally, by overexpressing betatrophin in the liver of mice, the authors recorded a 17-fold increase in beta cell proliferation, which lead to a threefold increase in beta cell mass. The betatrophin-overexpressing animals had a lower fasting glucose, a better glucose tolerance, and a normal insulin tolerance response.
Subsequent publications from the same and another lab have also reported on the stimulatory effects of betatrophin on murine beta cell proliferation with much more modest effects [32••, 42]. However, other preclinical studies have raised questions about betatrophin. Studies of betatrophin-deficient mice have shown that (1) they have a normal glucose tolerance [43•] and (2) their beta cells undergo normal expansion in response to insulin resistance caused by high-fat diet or from the administration of S961, thus questioning the importance of betatrophin for beta cell mass expansion [44••]. Furthermore, no increase in beta cell mass was observed when betatrophin was overexpressed in normal mice in two different studies, although an increase in triglyceride levels was observed [44••, 45••]. In the betatrophin-deficient mice, a decrease in body fat and triglyceride levels after feeding was instead recorded [43•]. Finally, thus far, a receptor for betatrophin on beta cells, or any other cell, has not been described.
There has yet been no documented effect of betatrophin on human beta cell proliferation or mass. In the publication by Jiao et al. [42], both human and mouse islets were transplanted under the kidney capsule of the mice, followed by treatment with the insulin receptor antagonist S961 to increase insulin resistance, which previously have been shown to also increase the hepatic expression of betatrophin. This lead to an increase in beta cell proliferation in both the native and transplanted mouse islets, but there was no effect on the transplanted human beta cells. Since human beta cell proliferation is highly age-dependent and primarily occurs in very young individuals [17], it is of interest to note that one of the five islet donors was 4 years of age, another 18 years, whereas the rest were above 40 years of age.
Betatrophin Levels in Humans Without Diabetes
To date, 12 studies have reported on betatrophin and its correlates in humans without diabetes (Table 1). The largest report on betatrophin levels in non-diabetic individuals (n = 1047) reported a positive association between betatrophin and age, BMI, waist/hip ratio, fasting plasma glucose, HbA1c, plasma levels of insulin, triglyceride levels, and homeostasis model assessment of insulin resistance (HOMA-IR) [46••].
In the first study on plasma betatrophin levels in humans, we reported that the levels of betatrophin in adult, healthy normal-weight individuals were positively associated with age, whereas we observed no association with fasting glucose levels, HbA1c, BMI, or blood lipids [47••]. This positive association between betatrophin and age among non-diabetic individuals has been confirmed in several studies [46••, 48•, 49, 50]. In a pediatric group, betatrophin levels were reported to be increased in children older than 8 years of age and also to be higher in male when compared to BMI-matched females [51].
The association between BMI and betatrophin levels in individuals without diabetes has been mixed across studies with one study showing a negative association between betatrophin and BMI [52] and others showing positive associations [46••, 53]. Consistent with what has been reported on the mRNA expression of betatrophin in rodents [38–40], the circulating levels of betatrophin have been found to be increased two hours after a defined meal in lean individuals [53].
The published data on betatrophin in healthy individuals are summarized in Table 1. All of the published studies so far are cross-sectional.
Betatrophin Levels in Patients With Insulin Resistance and Diabetes
In contrast to the animal model with acute loss of beta cells [32••], we found increased levels of betatrophin among patients with type 1 diabetes when compared to age- and BMI-matched healthy individuals [47••]; however, betatrophin levels did not differ between type 1 diabetes patients with detectable C-peptide levels compared to those without detectable C-peptide levels.
In the first report on betatrophin levels in patients with type 2 diabetes, there was no difference in betatrophin levels between those with and without type 2 diabetes [48•]. Following this report, there have been a number of reports showing increased levels of betatrophin in patients with type 2 diabetes [46••, 49, 50, 53, 54] and also reports on unaltered or decreased levels of betatrophin [52, 55]. The largest study on betatrophin levels in patients with type 2 diabetes (n = 556) reported increased betatrophin levels among those with type 2 diabetes vs. healthy controls [46••].
In the initial report on betatrophin by Melton’s group [32••], the expression of betatrophin mRNA levels in the liver was increased in the studied animal models of obesity and type 2 diabetes, ob/ob and db/db mice, and the expression increased as a response to induced insulin resistance by treatment with the insulin receptor antagonist S961. Furthermore, overexpression of betatrophin itself improved glycemic control. Based on these findings, it seems as if betatrophin itself does not induce insulin resistance but acts as a response in order to increase beta cell mass for the maintenance of normal glucose homeostasis. Therefore, if betatrophin plays the same or any role in humans, an increase of betatrophin would be expected to occur in insulin-resistant individuals in order to prevent the development of manifest diabetes. In the report by Chen et al. [50], while levels of betatrophin were found to be similar among healthy controls, individuals with impaired fasting glucose, and impaired glucose tolerance, betatrophin levels were increased in patients with newly diagnosed type 2 diabetes. Furthermore, it has been reported that the levels of betatrophin are positively associated with the duration of type 2 diabetes [46••, 56], although autopsy studies have demonstrated a decline in beta cell mass in long-standing type 2 diabetes [3, 4].
Cross-sectional studies do not demonstrate a better glycemic control (e.g., lower fasting blood glucose, HbA1c) in humans with type 2 diabetes with higher vs. lower betatrophin levels, and some studies even show that betatrophin levels are higher among those with worse glycemic control [49, 50, 53]. However, these data are biased by the different treatment regimens used in the different study populations, and the effect of the most commonly used anti-diabetic drugs on betatrophin levels are so far unknown.
In two reports, a positive association between betatrophin and HOMA-IR was observed [50, 55] but there are also reports on a negative association with HOMA-IR [54, 57, 58]. It should be mentioned though that in the latter reports, two are in patients with new onset type 2 diabetes [54, 57] and one in patients with type 2 diabetes onset in the young, in which the correlations was calculated for all study participants [58]. In a study comparing individuals with normal glucose tolerance to those with impaired glucose tolerance, no difference in betatrophin levels was observed [50]. However, up to date, there is no longitudinal study of changes in betatrophin levels during the development of insulin resistance and diabetes.
There are many possible explanations for the discrepancy in results between the different studies in humans, the most obvious one being the use of different ELISA kits which either detect full-length betatrophin (N-terminal) or total betatrophin (both full-length and C-terminal fragments) [59•]. So far, the ELISA kit detecting full-length betatrophin has been the most commonly used in studies [46••, 47••, 48•, 49, 50, 56, 60]. Whether full-length betatrophin or its C-terminal fragments exert biological effects is presently unknown. Conditions of sampling may also contribute to discrepancy in results, since there are effects of fasting or fed state on both the expression of betatrophin [38–40] and the circulating betatrophin levels in humans [53]. Moreover, differences in the handling of blood samples, time to separation of plasma/serum, as well as storing conditions could affect the results. Our experience is that both full-length and total betatrophin levels in plasma are most sensitive to freeze-thaw cycles. The published data on betatrophin levels in individuals with insulin resistance and type 2 diabetes are summarized in Table 2. All of the published studies so far are cross-sectional.
Betatrophin and Triglyceride Levels in Humans
In animal models, betatrophin has been tightly linked to triglyceride levels, and mice lacking betatrophin have significantly lower triglyceride levels than normal mice [43•] whereas overexpression of betatrophin leads to increased triglyceride levels [38, 44••, 45••]. In the studies on betatrophin levels in subjects with diabetes, there are no reports of an association between betatrophin and triglyceride levels. In two reports, a positive association with total cholesterol levels was observed [48•, 50], and in one report, an inverse association was observed [46••]. A possible confounding factor in the human studies is that many of the individuals could have been treated with lipid-lowering drugs which would affect triglyceride and possibly betatrophin levels.
Conclusion
The prevalence of type 2 diabetes is rapidly increasing worldwide, and currently, there are no therapies aiming to restore or even prevent the loss of beta cell mass. The discovery of betatrophin, a hormone that in the first study in mice counteracted glucose intolerance induced by insulin resistance by potently stimulating beta cell proliferation, boosted the hope to use this hormone as a drug for diabetes treatment. However, the findings have not been transferable to human beta cell proliferation to date, and the original findings in mice have also later been questioned. In humans, plasma betatrophin levels seem to increase with age, and to be increased in both patients with type 1 and type 2 diabetes. High betatrophin levels are generally not associated with better metabolic control, or preserved insulin production, and may in fact be associated with worse measures of these factors. There are many discrepancies in results between studies of betatrophin. Notably, studies of betatrophin levels in humans have been cross-sectional and therefore cannot address if betatrophin is a beneficial compensatory response to the changes that occur prior to the onset of diabetes. Also, differences in the techniques for measuring the hormone are probably a major contributing factor. Taken together, there is so far no evidence for betatrophin as a stimulator of human beta cell proliferation, and its potential importance in the pathophysiology of type 2 diabetes is presently unclear.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Lam DW, LeRoith D. The worldwide diabetes epidemic. Curr Opin Endocrinol Diabetes Obes. 2012;19(2):93–6.
Ritzel RA, Butler AE, Rizza RA, Veldhuis JD, Butler PC. Relationship between beta-cell mass and fasting blood glucose concentration in humans. Diabetes Care. 2006;29(3):717–8.
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–10.
Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10 Suppl 4:32–42.
Rahier J, Goebbels RM, Henquin JC. Cellular composition of the human diabetic pancreas. Diabetologia. 1983;24(5):366–71.
Clark A, Jones LC, de Koning E, Hansen BC, Matthews DR. Decreased insulin secretion in type 2 diabetes: a problem of cellular mass or function? Diabetes. 2001;50 Suppl 1:S169–71.
Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14(10):619–33.
Keenan HA, Sun JK, Levine J, et al. Residual insulin production and pancreatic ss-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes. 2010;59(11):2846–53.
Wang L, Lovejoy NF, Faustman DL. Persistence of prolonged C-peptide production in type 1 diabetes as measured with an ultrasensitive C-peptide assay. Diabetes Care. 2012;35(3):465–70.
Oram RA, Jones AG, Besser RE, et al. The majority of patients with long-duration type 1 diabetes are insulin microsecretors and have functioning beta cells. Diabetologia. 2014;57(1):187–91.
Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF. Human beta-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes. 2012;61(9):2205–13.
Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocana A. Human beta-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes. 2014;63(3):819–31.
Stewart AF, Hussain MA, Garcia-Ocana A, et al. Human beta-cell proliferation and intracellular signaling: part 3. Diabetes. 2015;64(6):1872–85.
Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, Garcia-Ocana A, Stewart AF. Diabetes mellitus—advances and challenges in human beta-cell proliferation. Nat Rev Endocrinol. 2015;11(4):201–12.
Gregg BE, Moore PC, Demozay D, et al. Formation of a human beta-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab. 2012;97(9):3197–206.
Meier JJ, Butler AE, Saisho Y, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57(6):1584–94.
Perl S, Kushner JA, Buchholz BA, et al. Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab. 2010;95(10):E234–9.
Menge BA, Tannapfel A, Belyaev O, et al. Partial pancreatectomy in adult humans does not provoke beta-cell regeneration. Diabetes. 2008;57(1):142–9.
Butler AE, Cao-Minh L, Galasso R, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia. 2010;53(10):2167–76.
Parnaud G, Bosco D, Berney T, et al. Proliferation of sorted human and rat beta cells. Diabetologia. 2008;51(1):91–100.
Banerjee M, Virtanen I, Palgi J, Korsgren O, Otonkoski T. Proliferation and plasticity of human beta cells on physiologically occurring laminin isoforms. Mol Cell Endocrinol. 2012;355(1):78–86.
Levitt HE, Cyphert TJ, Pascoe JL, et al. Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia. 2011;54(3):572–82.
Purwana I, Zheng J, Li X, et al. GABA promotes human beta-cell proliferation and modulates glucose homeostasis. Diabetes. 2014;63(12):4197–205.
Dirice E, Kahraman S, Jiang W, et al. Soluble factors secreted by T cells promote beta-cell proliferation. Diabetes. 2014;63(1):188–202.
In't Veld P, De Munck N, Van Belle K, et al. Beta-cell replication is increased in donor organs from young patients after prolonged life support. Diabetes. 2010;59(7):1702–8.
Grapensparr L, Vasylovska S, Li Z, et al. Co-transplantation of human pancreatic islets with post-migratory neural crest stem cells increases beta-cell proliferation and vascular and neural regrowth. J Clin Endocrinol Metab. 2015;100(4):E583–90.
Takane KK, Kleinberger JW, Salim FG, Fiaschi-Taesch NM, Stewart AF. Regulated and reversible induction of adult human beta-cell replication. Diabetes. 2012;61(2):418–24.
Fiaschi-Taesch N, Bigatel TA, Sicari B, et al. Survey of the human pancreatic beta-cell G1/S proteome reveals a potential therapeutic role for cdk-6 and cyclin D1 in enhancing human beta-cell replication and function in vivo. Diabetes. 2009;58(4):882–93.
Fiaschi-Taesch NM, Salim F, Kleinberger J, et al. Induction of human beta-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes. 2010;59(8):1926–36.
Imai J, Katagiri H, Yamada T, et al. Regulation of pancreatic beta cell mass by neuronal signals from the liver. Science. 2008;322(5905):1250–4.
Olerud J, Mokhtari D, Johansson M, et al. Thrombospondin-1: an islet endothelial cell signal of importance for {beta}-cell function. Diabetes. 2011;60(7):1946–54.
Yi P, Park JS, Melton DA. Betatrophin: a hormone that controls pancreatic beta cell proliferation. Cell. 2013;153(4):747–58. The publication in which betatrophin and its effects on beta cell proliferation in mice was first described.
Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA. Butler PC: beta-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36(1):111–7.
Sullivan BA, Hollister-Lock J, Bonner-Weir S, Weir GC. Reduced Ki67 staining in the postmortem state calls into question past conclusions about the lack of turnover of adult human beta-cells. Diabetes. 2015;64(5):1698–702.
Tyrberg B, Ustinov J, Otonkoski T, Andersson A. Stimulated endocrine cell proliferation and differentiation in transplanted human pancreatic islets: effects of the ob gene and compensatory growth of the implantation organ. Diabetes. 2001;50(2):301–7.
Gargani S, Thevenet J, Yuan JE, et al. Adaptive changes of human islets to an obesogenic environment in the mouse. Diabetologia. 2013;56(2):350–8.
Dong XY, Pang XW, Yu ST, et al. Identification of genes differentially expressed in human hepatocellular carcinoma by a modified suppression subtractive hybridization method. Int J Cancer. 2004;112(2):239–48.
Zhang R. Lipasin, a novel nutritionally-regulated liver-enriched factor that regulates serum triglyceride levels. Biochem Biophys Res Commun. 2012;424(4):786–92.
Ren G, Kim JY, Smas CM. Identification of RIFL, a novel adipocyte-enriched insulin target gene with a role in lipid metabolism. Am J Physiol Endocrinol Metab. 2012;303(3):E334–51.
Quagliarini F, Wang Y, Kozlitina J, et al. Atypical angiopoietin-like protein that regulates ANGPTL3. Proc Natl Acad Sci U S A. 2012;109(48):19751–6.
Fu Z, Yao F, Abou-Samra AB, Zhang R. Lipasin, thermoregulated in brown fat, is a novel but atypical member of the angiopoietin-like protein family. Biochem Biophys Res Commun. 2013;430(3):1126–31.
Jiao Y, Le Lay J, Yu M, Naji A, Kaestner KH. Elevated mouse hepatic betatrophin expression does not increase human beta-cell replication in the transplant setting. Diabetes. 2014;63(4):1283–8.
Wang Y, Quagliarini F, Gusarova V, et al. Mice lacking ANGPTL8 (Betatrophin) manifest disrupted triglyceride metabolism without impaired glucose homeostasis. Proc Natl Acad Sci U S A. 2013;110(40):16109–14. The authors have investigated mice lacking betatrophin and found no negative effect on glucose homeostasis.
Gusarova V, Alexa CA, Na E, et al. ANGPTL8/betatrophin does not control pancreatic beta cell expansion. Cell. 2014;159(3):691–6. Gusarova et al. found that mice lacking betatrophin have a normal development of beta cell mass and a normal response and increase in beta cell proliferation in response to insulin resistance which puts the relevance of betatrophin into question.
Cox AR, Lam CJ, Bonnyman CW, Chavez J, Rios JS, Kushner JA. Angiopoietin-like protein 8 (ANGPTL8)/betatrophin overexpression does not increase beta cell proliferation in mice. Diabetologia. 2015;58(7):1523–31. The authors have overexpressed betatrophin but do not find any increase in beta cell proliferation or beta cell mass which questions the findings of Ye et al. [32].
Abu-Farha M, Abubaker J, Al-Khairi I, et al. Higher plasma betatrophin/ANGPTL8 level in Type 2 Diabetes subjects does not correlate with blood glucose or insulin resistance. Sci Rep. 2015;5:10949. The largest study so far on circulating betatrophin levels in healthy- and individuals with type 2 diabetes. The betatrophin levels were found to be increased in type 2 diabetes.
Espes D, Lau J, Carlsson PO. Increased circulating levels of betatrophin in individuals with long-standing type 1 diabetes. Diabetologia. 2014;57(1):50–3. The first publication on circulating betatrophin levels in plasma in healthy individuals and individuals with type 1 diabetes. In contrast to the animal model we found the levels of betatrophin to be increased in patients with type 1 diabetes.
Fenzl A, Itariu BK, Kosi L, et al. Circulating betatrophin correlates with atherogenic lipid profiles but not with glucose and insulin levels in insulin-resistant individuals. Diabetologia. 2014;57(6):1204–8. The first publication on circulating betatrophin levels in patients with type 2 diabetes, no difference in betatrophin levels between healthy- and individuals with type 2 diabetes was observed.
Espes D, Martinell M, Carlsson PO. Increased circulating betatrophin concentrations in patients with type 2 diabetes. Int J Endocrinol. 2014;2014:323407.
Chen X, Lu P, He W, et al. Circulating betatrophin levels are increased in patients with type 2 diabetes and associated with insulin resistance. J Clin Endocrinol Metab. 2015;100(1):E96–100.
Wu S, Gao H, Ma Y, Fu L, Zhang C, Luo X. Characterisation of betatrophin concentrations in childhood and adolescent obesity and insulin resistance. Pediatr Diabetes. 2014.
Gomez-Ambrosi J, Pascual E, Catalan V, et al. Circulating betatrophin concentrations are decreased in human obesity and type 2 diabetes. J Clin Endocrinol Metab. 2014;99(10):E2004–9.
Fu Z, Berhane F, Fite A, Seyoum B, Abou-Samra AB, Zhang R. Elevated circulating lipasin/betatrophin in human type 2 diabetes and obesity. Sci Rep. 2014;4:5013.
Hu H, Sun W, Yu S, et al. Increased circulating levels of betatrophin in newly diagnosed type 2 diabetic patients. Diabetes Care. 2014;37(10):2718–22.
Guo K, Lu J, Yu H, et al. Serum betatrophin concentrations are significantly increased in overweight but not in obese or type 2 diabetic individuals. Obesity (Silver Spring). 2015;23(4):793–7.
Tokumoto S, Hamamoto Y, Fujimoto K, et al. Correlation of circulating betatrophin concentrations with insulin secretion capacity, evaluated by glucagon stimulation tests. Diabet Med. 2015;32(5):653–6.
Xie X, Gao T, Yang M, et al. Associations of betatrophin levels with irisin in Chinese women with normal glucose tolerance. Diabetol Metab Syndr. 2015;7:26.
Gokulakrishnan K, Manokaran K, Pandey GK, et al. Relationship of betatrophin with youth onset type 2 diabetes among Asian Indians. Diabetes Res Clin Pract. 2015;109(1):71–6.
Fu Z, Abou-Samra AB, Zhang R. An explanation for recent discrepancies in levels of human circulating betatrophin. Diabetologia. 2014;57(10):2232–4. This study describe the different ELISA kits used to measure betatrophin which explain some of the differences in the published levels and correlations of betatrophin.
Yamada H, Saito T, Aoki A, et al. Circulating betatrophin is elevated in patients with type 1 and type 2 diabetes. Endocr J. 2015;62(5):417–21.
Acknowledgments
The original research cited from our group was generously supported by the Swedish Research Council (K2013-55X-15043), the EFSD/JDRF/Novo Nordisk Programme 2012, AFA Insurance, the Swedish Diabetes Association, the Swedish Juvenile Diabetes Foundation, Olle Engkvist Byggmästare Foundation, and the Novo Nordisk Foundation and Diabetes Wellness Sverige.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
Daniel Espes, Mats Martinell, Hanna Liljebäck, and Per-Ola Carlsson have no conflict of interest to declare.
Human and Animal Rights and Informed Consent
This article does not contain any original studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Diabetes Epidemiology
Rights and permissions
About this article

Cite this article
Espes, D., Martinell, M., Liljebäck, H. et al. Betatrophin in Diabetes Mellitus: the Epidemiological Evidence in Humans. Curr Diab Rep 15, 104 (2015). https://doi.org/10.1007/s11892-015-0676-4
Published:
DOI: https://doi.org/10.1007/s11892-015-0676-4