
Abstract
Type 2 diabetes (T2D) is a chronic non-communicable disease that is driven by insulin resistance as a result of increasing obesity and decreasing activity levels that occur with increasing age. This disease generally develops after the age of 40, but it is now increasingly diagnosed in children and young adults. Increasing evidence, however, suggests that T2D can originate during early development. It has been repeatedly found that malnutrition during the gestational period can result in intrauterine growth restriction and low birth weight, which in combination with postnatal catch-up growth may subsequently lead to the development of T2D. There is ample evidence that T2D may also be programmed by maternal substance abuse (the harmful use of psychoactive substances such as illicit drugs or alcohol) during pregnancy and/or lactation. The research activity in this field is currently mainly focused on the childhood health problems following prenatal exposures to substance abuse. The delayed programming effects on adult-onset disorders, including metabolic syndrome and T2D, however, have been reported only rarely. This review provides animal and human evidence that early-life exposure to substance abuse, including alcohol, nicotine, and cocaine, may program not only childhood health outcomes but also life-long metabolic health status, including risk of T2D and related conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes (T2D) is a chronic non-communicable disease that is driven by insulin resistance as a result of increasing obesity and decreasing activity levels that occur with increasing age. This disease generally develops after the age of 40, but it is now increasingly diagnosed in children and young adults [1]. There is increasing experimental and epidemiological evidence that T2D and obesity, which is an important risk factor for T2D, may originate during critical windows of prenatal and early postnatal development [2]. These findings are consistent with the developmental programming of health and disease (DOHAD) hypothesis which proposes that physiology and structure of the developing fetus may be adapted in response to adverse developmental conditions, such as poor nutrition, predisposing to various pathological conditions later in life [3]. In the case of T2D, fetus likely can be adapted to poor nutritional environment in a way to reduce capacity to produce insulin and increase insulin resistance thus providing short-term survival benefit but predisposing to the development of T2D in conditions of postnatal food abundance. The precise mechanisms underlying developmental programming of obesity and T2D are far from being completely elucidated. However, recent studies suggest that epigenetics (heritable changes in gene function that do not involve changes to the DNA sequence) is the most plausible mechanistic pathway for the relationship between adverse developmental conditions and health outcomes in later life [2].
Maternal malnutrition and gestational diabetes were shown to be the most important contributors to obesity and metabolic dysfunction in offspring [3]. A relation between birth weight, which is an indicator of fetal growth, and later-life risk of T2D was shown to be not linear but rather U-shaped, and high birth weight is associated with an increased risk of T2D to the same extent as low birth weight [4]. Along with malnutrition, substance abuse (the harmful use of psychoactive substances such as illicit drugs or alcohol) during the prenatal developmental stages is likely another important determinant of the process of developmental programming in modern human beings. Substance abuse is currently among the major public health concerns facing many nations around the world due to its high prevalence and long-term health consequences for the next generations [5]. Presently, the research activity in this field is mainly focused on the childhood and adolescent outcomes, primarily cognitive, neurophysiological, neurological, and psychosocial problems, following prenatal exposures to substance abuse [6, 7•]. The delayed programming effects on aging-associated diseases, including metabolic disorders, however, have been reported only rarely.
This review aimed to provide the evidence that exposure to substance abuse in early life may program not only health outcomes during childhood but also life-long health status, including risk of T2D and related conditions in adulthood.
Tobacco Smoking
Nicotine is a psychoactive plant alkaloid responsible for the addictive properties of tobacco. Current statistics indicate that, in spite of medical advice, 20–30 % of smoking women continue to smoke during pregnancy, representing about 10 % of all pregnancies [8]. Tobacco smoking during pregnancy was repeatedly shown to be associated with a number of adverse effects on in utero development, including intrauterine growth restriction (IUGR) and shortened gestation [9]. The effects of maternal smoking can, however, be extended beyond the developmental period and affect health status in adulthood and later life [8, 10••]. An association between a maternal exposure to nicotine during pregnancy and lactation and different aspects of metabolic syndrome in offspring, including altered glucose homeostasis, increased blood pressure, and obesity, has been demonstrated in rat studies [11]. The hypertrophy of adipocytes, leptin and insulin resistance, as well as thyroid and adrenal dysfunction in adult life were observed in rodents exposed to nicotine during the lactation period only [12, 13]. Remarkably, the consequences of early-life exposure to nicotine have been shown to be not limited to the first generation but were also manifested in the second-generation offspring who exhibited hypertension, enhanced levels of fasting serum insulin, and elevated insulin response to oral glucose load [14].
In humans, it has been repeatedly shown that nicotine exposure in utero may cause adverse neonatal outcomes [15]. In particular, it may influence the birth weight which is a good predictor of metabolic health in adult life [16]. For example, in examining data on 366,886 Danish singletons, it has been revealed that both birth weight and abdominal circumference decreased with maternal smoking [17]. Data from several studies also indicate that maternal smoking during pregnancy may cause an elevated risk of hypertension, obesity, and diabetes, as well as cholesterol development along with a tendency towards a detrimental lipoprotein profile during childhood [18–20]. In a meta-analysis of 14 observational studies (n = 84,563), odds of having overweight or obese children at 3–33 years of age were 1.5 times greater in mothers who smoked during pregnancy compared to those who had never smoked [21].
A causal relationship between prenatal exposure to nicotine and various aspects of metabolic syndrome and T2D in adulthood was also reported in several studies [10••, 22–24]. In the Norwegian Mother and Child Cohort Study (MoBa), including 74,023 pregnancies from 1999 to 2008, after adjusting for age, education, and personal smoking, the adjusted odds of obesity, hypertension, and T2D in women 14–47 years of age who were exposed to tobacco smoke in utero were 1.53, 1.68, and 1.14, respectively, compared to those in unexposed women [10••]. In several studies, the relationship between maternal smoking during pregnancy and cardiovascular complications, which are known to be associated with metabolic syndrome [25], has been found. The most significant findings were reported in the 1958 British birth cohort study [22–24]. Remarkably, in terms of causal explanation, that offspring of mothers who smoked during pregnancy had, in this cohort, a lower birth weight than offspring of non-smoking mothers, but from adolescence, they demonstrated a tendency to have an increase of BMI with age [22]. At age 33, the ORs for obesity associated with maternal smoking were 1.41 and 1.56 for women and men, respectively. These findings were robust to adjustment for early-life, childhood, and adulthood factors [22]. After adjustment for sex and family history of T2D, maternal smoking during pregnancy was associated with glycated hemoglobin (HbA1c) levels at 45 years of age (OR = 1.33) [23]. Offspring of smoking mothers were shown to be more likely to have a more adverse cardiovascular risk profile in mid-adulthood which is manifested by elevated levels of blood pressure, HbA1c, and triglycerides, as well as by higher adult BMI and waist circumference, compared to offspring of non-smoker mothers [24]. These associations, however, were abolished, except for BMI and waist circumference, after adjustment for postnatal influences across the life span. The association with exposure to nicotine during gestation was observed also for vascular damage such as common carotid artery intima-media thickness in young adulthood [26]. In the British longitudinal birth cohort, those persons who were born to medium, variable, and heavy smoking mothers had, after adjustment for sex, mother’s age at birth of cohort member, age mother left school, family social class at birth, birth weight, own smoking at age 16 years, and BMI at age 33 years, 1.11, 4.13, and 4.55 times higher odds to have diabetes between 16 and 33 years of age, respectively, compared to non-smoking mothers [27••]. The corresponding ORs for obesity associated with maternal smoking during pregnancy were 1.34, 1.35, and 1.38, with a statistically significant trend for medium, variable, and heavy smokers, respectively. More recently, similar findings were obtained in research using data from the Swedish Medical Birth Register [28•]. After adjustment for maternal parity, age at pregnancy and mode of delivery, as well as for personal smoking, those daughters who were moderately or heavily exposed to maternal smoking in utero had 1.62 and 1.52 times higher odds to have gestational diabetes, respectively, than daughters of non-smoking mothers. The corresponding adjusted ORs for obesity were 1.36 and 1.58. In US Nurses’ Health Study II [29], it has been found that after adjustment for behavioral and socioeconomic covariates, prenatal exposure to maternal smoking was associated with adiposity at the ages of 5–10, 18, and in adulthood. The ORs for obesity during adulthood were 1.26, 1.46, and 1.43 for those daughters whose mothers smoked 1–14, 15–24, and 25+ cigarettes per day, respectively, compared to offspring of non-smoking mothers. Interestingly, women whose fathers smoked during their intrauterine life also had an elevated risk of being overweight and obese during adulthood [29]. The relationship between secondhand smoking in childhood and adolescent periods and T2D in adulthood was revealed in a prospective French cohort study (n = 37,343), where it has been demonstrated that women, who had at least one smoking parent during their childhood and adolescence, had subsequently 18 % higher odds to have T2D than women with never-smoking parents [30].
Alcohol
Moderate alcohol consumption is known to reduce the risk of developing T2D; chronic heavy alcohol consumption, however, is a prospective risk factor for the development of this disease [31]. Alcohol consumed during pregnancy can cross the placenta and indirectly change fetal development by disrupting the hormonal interactions between the mother, placenta, and fetus [32]. Heavy exposure to alcohol during gestational period is well-known to lead to a variety of developmental anomalies, low birth weight, and severely impair neurobehavioral and physical development, causing fetal alcohol spectrum disorders (FASD) [33]. However, despite this knowledge, 12 % of women continue to drink alcohol during pregnancy [34]. Long-term neurobehavioral consequences of prenatal alcohol exposure have been reported repeatedly [35]; lasting metabolic outcomes, though not yet thoroughly studied in humans, have been repeatedly observed in animal models.
In rats, intrauterine exposure to alcohol was found to cause long-term reduction in plasma levels of insulin-like growth factor 1 (IGF1) and IGF-binding proteins [36]. This finding is important from the etiological standpoint since IGF1 pathway is known to be substantially involved in glucose metabolism, and both decreased or elevated levels of IGF1 are shown to increase the risk of T2D [37]. The causal link between maternal dietary alcohol consumption and risk of metabolic complications during adulthood was evident from the data obtained in the study by Pennington et al. [38], where exposure to alcohol during in utero development resulted in hypertriglyceridemia along with an increase in the very low-density lipoprotein fraction of serum, both known to be associated with metabolic syndrome and T2D, in adult rat offspring. The links between maternal alcohol exposure during pregnancy and beta cell dysfunction, abnormal glucose homeostasis, and insulin resistance during adulthood were evident in a line of studies [39–43]. In addition, insulin resistance was also evident after exposure to alcohol during lactation [42]. Prenatal exposure to alcohol resulted in insulin resistance along with enhanced expression levels of hepatic gluconeogenic genes, explaining the increased gluconeogenesis in adult rats, in the Yao et al. [44] study. Prenatal alcohol exposure has been shown to increase susceptibility to high-fat diet-induced MS in adult male rat offspring [45••], as well as to high-fat diet-induced non-alcoholic fatty liver disease (a condition known to enhance the risk of the development of T2D [46]) in adult female offspring rats [47]. Remarkably, the programming effects of alcohol consumption during pregnancy were shown to be able to persist across generations. In the study by Harper et al. [48], the effects of grandmaternal exposure to alcohol on insulin and functional glucose responses in the second-generation offspring were observed, possibly owing to the effect of alcohol on the germ line of the F1 fetus.
The evidence for programming effects of prenatal alcohol exposure was also obtained in guinea pig which is a commonly used model system in research of teratogenicity of alcohol because its prenatal development is much more similar to the humans relative to other rodent models [49]. In this animal model, increased whole-body and pancreatic adiposity was observed in the offspring of dams exposed to alcohol during pregnancy [49]. More recently, the same authors extended their findings by showing that exposure to alcohol in utero alters both peripheral and central expression of insulin/IGF signaling molecules at the level of mRNA, which can be linked to metabolic dysregulation in offspring during adulthood [50].
To date, human evidence describing the long-term effects of alcohol exposure is scarce. By using the life course model of self-rated health, Bauldry et al. [51] revealed that both parental alcoholism and smoking can predict obesity in offspring, and this association was strengthened with offspring age.
Cocaine
According to the Substance Abuse and Mental Health Services Administration (SAMHSA) survey conducted in the USA, nearly 5 % of women reported illicit drug use, including cocaine, during their pregnancy [52]. Infants born to mothers who used cocaine throughout pregnancy have various health problems including low birth weight and abnormalities of different systems such as lungs, genitals, liver, and neurological system [53, 54], as well as a number of cardiac malformations [55]. Exposure to cocaine in utero was shown to be linked to a variety of neurodevelopmental, behavioral, and cognitive problems and also to numerous cardiovascular complications in childhood [56]. Taking the latest into account, it can be assumed that prenatal cocaine exposure may be linked to cardiometabolic risk factors, such as hypertension, increased levels of C-reactive protein and lipids, and insulin resistance in later life [57]. Evidence for the link between prenatal cocaine exposure and metabolic disturbances in later childhood associated with risk for subsequent development of T2D was obtained in research by La Gasse and co-authors who reported elevated blood pressure and BMI in 9-year-old children whose mothers consumed cocaine during pregnancy [58]. Prenatally cocaine-exposed children were also four times as likely to become obese at 9 years of age if they were not exposed to alcohol as well [59]. These findings suggest that alcohol exposure can attenuate the effect of exposure to cocaine during gestation on BMI and obesity. If this is true, then failure to demonstrate a link between prenatal cocaine exposure and later-life obesity could be in part be due to the common practice of consuming both these drugs together throughout the pregnancy.
Summarizing, it should be emphasized that, despite the high rate of consumption of cocaine, alcohol, and tobacco in pregnancy and importance of this problem for the public health care, epidemiological studies of the long-term effects of exposure to these substances in early life on the risk of T2D in subsequent life are scarce until now. The research of other abused substances such as amphetamine, marijuana, opioids, etc., had never been realized until now in this area. The potential risks of long-lasting metabolic consequences of early-life exposure to these psychoactive drugs remain to be evaluated in further research.
Mechanisms Linking Early-Life Exposure to Substance Abuse to Later-Life Metabolic Disorders
Currently, it is widely accepted that IUGR caused by maternal undernutrition, placental insufficiency, hypoxia, infections, etc. increases the susceptibility to metabolic complications, including T2D, later in life. However, not linearly inverse but a U-shaped relationship between birth weight and T2D risk is evident in most studies, and both small-for-gestational-age (SGA) and large-for-gestational-age (LGA) infants are shown to be at risk for subsequent development of T2D [4]. Maternal substance abuse in pregnancy most commonly leads to IUGR/SGA conditions because fetus in these circumstances is occasionally depleted in nutrients, suggesting episodic exposure to fetal malnutrition [60]. Depletion in oxygen and hypoxic stress are also possible. For example, the rhesus monkey study showed that, penetrating through the placenta, tobacco smoke metabolites may act as vasoconstrictors reducing uterine blood flow up to 38 % [61]. A risk for developing IUGR was reported repeatedly for different substances of abuse, including nicotine, alcohol, cannabis, and opiates, as well as for poly-drug abuse [9, 62–65].
All substances of abuse are known to disrupt eating behavior and cause nutrient deficiencies and malnutrition [66]. Reduced appetite and decreased food intake have been demonstrated for abuse of alcohol [67], nicotine [68], and cocaine [69]. Thereby, the appetite dysregulation appears to be a crucial contributing factor in substance abuse-induced IUGR. The important point seems to be that withdrawal of substance use such as nicotine [70], cocaine [71], and opiates [72] causes stimulation of appetite. In fact, if child receives substance of abuse from the mother through the placenta or in breast milk, but after weaning no longer receives that, the same mechanism may be operative, leading subsequently to the risk of overeating, obesity, and T2D. This may likely explain the phenomenon of accelerated postnatal weight gain and high prevalence of obesity reported among offspring of mothers who smoked during pregnancy [73].
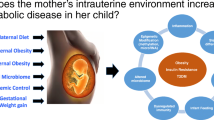
Developmental programming of hypothalamic regulation of appetite and adipogenic signals controlling lipogenesis has been proposed as a key mechanism underlying development of obesity and T2D in offspring [74]. Such effects are most studied for maternal tobacco smoking. The major reported effects of prenatal nicotine exposure include (1) high rate of beta cell apoptosis, reduced size and number of islets in the pancreas, and impaired glucose homeostasis and (2) promotion of adipocyte differentiation and adipogenesis, both processes known to be involved in obesity and the progression of T2D [8]. Moreover, it is commonly believed that epigenetic regulation of the expression of transcription factors regulating glucose and lipid metabolism plays a key role in programming effects of prenatal exposure to substance abuse [35]. In addition, the impairment of the fetal vessel flow has been proposed as underlying mechanisms [9]. A hypothetical mechanism linking prenatal exposure to substance of abuse to development of metabolic syndrome and T2D in adult life is presented in Fig. 1.

A hypothetical scheme of the main mechanisms linking prenatal exposure to substance of abuse to development of metabolic syndrome and T2D in adult life
Conclusions
A trend to a dramatic increase in the incidence of T2D is becoming increasingly evident across both developed and developing countries. Metabolic syndrome and related complications, such as elevated blood pressure, dyslipidemia, impaired glucose metabolism, and T2D, are among the major causes of death in western societies. Consistent findings have recently been reported, suggesting that risk for T2D may be programmed in early development. Currently, maternal malnutrition is considered to be a major factor involved in early-life programming of the metabolic syndrome and T2D. Another potentially important programming factor such as the maternal substance abuse during pregnancy has received less attention until recently, even though its long-lasting health effects are comparable to those of nutritional inadequacy in early life.
It is increasingly clear that substance abuse during pregnancy is among the important factors contributing to the current epidemic of T2D around the globe. The current increasing rates of obesity and T2D are likely caused not only by adult lifestyle changes in both developing and developed countries, such as westernized dietary habits and physical inactivity, but also by inadequate conditions, including malnutrition and stress, as well as exposure to substance abuse in early life. Adverse conditions during developmental period may trigger metabolic adaptations advantageous in early life but predisposing to chronic disease, including T2D, in adulthood [75]. Therefore, it is clear that research in this area is extremely important for public health and preventive medicine. However, despite the importance to health policy, research in this field is fairly limited. An increased risk of various metabolic complications in the offspring perinatally exposed to substances of abuse, including alcohol, nicotine, and cocaine, has been reported in several experimental and epidemiological studies, while impacts of other potentially dangerous psychoactive substances, such as opioids, amphetamine, marijuana, etc., have not been clarified so far. Unlike studies focused on neurophysiological and psychosocial problems triggered by early-life exposures to substances of abuse, where mechanistic links with exposures have been sufficiently clarified [6, 7•, 35], the research of the metabolic consequences of such exposures is still predominantly descriptive; the mechanistic links remain hypothetical. In this review, it has been speculated that appetite dysregulation may play a key role in linking early-life exposure to substance abuse and later risk for T2D. The questions on the mechanistic basis for this link remain, however, largely unanswered and should be addressed in future research. Advances in the understanding of mechanisms linking prenatal exposure to substance abuse to the risk of development of T2D in adulthood might provide the scientific basis for novel prevention and treatment paradigms.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Wilmot E, Idris I. Early onset type 2 diabetes: risk factors, clinical impact and management. Ther Adv Chronic Dis. 2014;5(6):234–44.
Vaag A, Brøns C, Gillberg L, Hansen NS, Hjort L, Arora GP, et al. Genetic, nongenetic and epigenetic risk determinants in developmental programming of type 2 diabetes. Acta Obstet Gynecol Scand. 2014;93(11):1099–108.
Inadera H. Developmental origins of obesity and type 2 diabetes: molecular aspects and role of chemicals. Environ Health Prev Med. 2013;18(3):185–97.
Harder T, Rodekamp E, Schellong K, Dudenhausen JW, Plagemann A. Birth weight and subsequent risk of type 2 diabetes: a meta-analysis. Am J Epidemiol. 2007;165(8):849–57.
Narkowicz S, Płotka J, Polkowska Ż, Biziuk M, Namieśnik J. Prenatal exposure to substance of abuse: a worldwide problem. Environ Int. 2013;54:141–63.
Sithisarn T, Granger DT, Bada HS. Consequences of prenatal substance use. Int J Adolesc Med Health. 2012;24(2):105–12.
Behnke M, Smith VC, Committee on Substance Abuse, Committee on Fetus and Newborn. Prenatal substance abuse: short- and long-term effects on the exposed fetus. Pediatrics. 2013;131(3):e1009–24. This review provides information on the short- and long-term effects of the most common drugs involved in prenatal exposure: alcohol, nicotine, cocaine, opiates, marijuana, and methamphetamine.
Somm E, Schwitzgebel VM, Vauthay DM, Aubert ML, Hüppi PS. Prenatal nicotine exposure and the programming of metabolic and cardiovascular disorders. Mol Cell Endocrinol. 2009;304:69–77.
Milnerowicz-Nabzdyk E, Bizoń A. Effect of cigarette smoking on vascular flows in pregnancies complicated by intrauterine growth restriction. Reprod Toxicol. 2014;50:27–35.
Cupul-Uicab LA, Skjaerven R, Haug K, Melve KK, Engel SM, Longnecker MP. In utero exposure to maternal tobacco smoke and subsequent obesity, hypertension, and gestational diabetes among women in the MoBa cohort. Environ Health Perspect. 2012;120:355–60. This study shows that exposure to nicotine in utero is associated with hypertension, obesity, and gestational diabetes mellitus in adult women.
Bruin JE, Gerstein HC, Holloway AC. Long-term consequences of fetal and neonatal nicotine exposure: a critical review. Toxicol Sci. 2010;116:364–74.
Freitas AL, Abreu-Villaça Y, Passos MC, Lisboa PC. Neonatal nicotine exposure causes insulin and leptin resistance and inhibits hypothalamic leptin signaling in adult rat offspring. J Endocrinol. 2010;206:55–63.
Lisboa PC, de Oliveira E, de Moura EG. Obesity and endocrine dysfunction programmed by maternal smoking in pregnancy and lactation. Front Physiol. 2012;3:437.
Holloway AC, Cuu DQ, Morrison KM, Gerstein HC, Tarnopolsky MA. Transgenerational effects of fetal and neonatal exposure to nicotine. Endocrine. 2007;31:254–9.
Rhee KE, Phelan S, McCaffery J. Early determinants of obesity: genetic, epigenetic, and in utero influences. Int J Pediatr. 2012;2012:9.
Alexander BT, Henry Dasinger J, Intapad S. Effect of low birth weight on women’s health. Clin Ther. 2014;36(12):1913–23.
Tanvig M. Offspring body size and metabolic profile—effects of lifestyle intervention in obese pregnant women. Dan Med J. 2014;61(7):B4893.
Von K, Toschke A, Koletzko B, Slikker W. Maternal smoking during pregnancy and childhood obesity. Am J Epidemiol. 2002;156:954–61.
Wideroe M, Vik T, Jacobsen G, Bakketeig L. Does maternal smoking during pregnancy cause childhood overweight? Paediatr Perinat Epidemiol. 2003;17:171–9.
Jaddoe VW, de Ridder MA, van den Elzen AP, Hofman A, Uiterwaal CS, Witteman JC. Maternal smoking in pregnancy is associated with cholesterol development in the offspring: a 27-years follow-up study. Atherosclerosis. 2008;196:42–8.
Oken E, Levitan EB, Gillman MW. Maternal smoking during pregnancy and child overweight: systematic review and meta-analysis. Int J Obes (Lond). 2008;32(2):201–10.
Power C, Jefferis BJ. Fetal environment and subsequent obesity: a study of maternal smoking. Int J Epidemiol. 2002;31 Suppl 2:413–9.
Thomas C, Hypponen E, Power C. Prenatal exposures and glucose metabolism in adulthood: are effects mediated through birth weight and adiposity? Diabetes Care. 2007;30:918–24.
Power C, Atherton K, Thomas C. Maternal smoking in pregnancy, adult adiposity and other risk factors for cardiovascular disease. Atherosclerosis. 2010;211:643–8.
Chiolero A, Kaufman JS. Metabolic mediators of body-mass index and cardiovascular risk. Lancet. 2014;383(9934):2042.
Geerts CC, Bots ML, Grobbee DE, Uiterwaal CS. Parental smoking and vascular damage in young adult offspring: is early life exposure critical? The atherosclerosis risk in young adults study. Arterioscler Thromb Vasc Biol. 2008;28:2296–302.
Montgomery SM, Ekbom A. Smoking during pregnancy and diabetes mellitus in a British longitudinal birth cohort. Br Med J. 2002;324:26–7. The hypothesis that maternal smoking during pregnancy increases both the risk of early onset type 2 diabetes and non-diabetic obesity in offspring was tested.
Mattsson K, Källén K, Longnecker MP, Rignell-Hydbom A, Rylander L. Maternal smoking during pregnancy and daughters’ risk of gestational diabetes and obesity. Diabetologia. 2013;56(8):1689–95. This study provides direct evidence that women prenatally exposed to nicotine are at higher risk of developing gestational diabetes and obesity.
Harris HR, Willett WC, Michels KB. Parental smoking during pregnancy and risk of overweight and obesity in the daughter. Int J Obes (Lond). 2013;37(10):1356–63.
Lajous M, Tondeur L, Fagherazzi G, de Lauzon-Guillain B, Boutron-Ruaualt MC, Clavel-Chapelon F. Childhood and adult secondhand smoke and type 2 diabetes in women. Diabetes Care. 2013;36(9):2720–5.
Pietraszek A, Gregersen S, Hermansen K. Alcohol and type 2 diabetes. A review. Nutr Metab Cardiovasc Dis. 2010;20(5):366–75.
Weinberg J, Sliwowska JH, Lan N, Hellemans KG. Prenatal alcohol exposure: foetal programming, the hypothalamic-pituitary-adrenal axis and sex differences in outcome. J Neuroendocrinol. 2008;20:470–88.
Dörrie N, Föcker M, Freunscht I, Hebebrand J. Fetal alcohol spectrum disorders. Eur Child Adolesc Psychiatry. 2014;23(10):863–75.
Pruett D, Waterman EH, Caughey AB. Fetal alcohol exposure: consequences, diagnosis, and treatment. Obstet Gynecol Surv. 2013;68(1):62–9.
Vaiserman AM. Long-term health consequences of early-life exposure to substance abuse: an epigenetic perspective. J DOHaD. 2013;4:269–79.
Breese CR, D’Costa A, Ingram RL, et al. Long-term suppression of insulin-like growth factor-1 in rats after in utero ethanol exposure: relationship to somatic growth. J Pharmacol Exp Ther. 1993;264 Suppl 1:448–56.
Schneider HJ, Friedrich N, Klotsche J, Schipf S, Nauck M, Völzke H, et al. Prediction of incident diabetes mellitus by baseline IGF1 levels. Eur J Endocrinol. 2011;164(2):223–9.
Pennington JS, Shuvaeva TI, Pennington SN. Maternal dietary ethanol consumption is associated with hypertriglyceridemia in adult rat offspring. Alcohol Clin Exp Res. 2002;26(6):848–55.
Minuk GY, Meyers AF, Legare DJ, Sadri P, Lautt WW. Fetal exposure to alcohol results in adult insulin resistance in the rat. Proc West Pharmacol Soc. 1998;41:39–40.
Elton CW, Pennington JS, Lynch SA, Carver FM, Pennington SN. Insulin resistance in adult rat offspring associated with maternal dietary fat and alcohol consumption. J Endocrinol. 2002;173(1):63–71.
Chen L, Nyomba BL. Effects of prenatal alcohol exposure on glucose tolerance in the rat offspring. Metabolism. 2003;52(4):454–62.
Chen L, Nyomba BL. Whole body insulin resistance in rat offspring of mothers consuming alcohol during pregnancy or lactation: comparing prenatal and postnatal exposure. J Appl Physiol (1985). 2004;96(1):167–72.
Yao XH, Grégoire Nyomba BL. Abnormal glucose homeostasis in adult female rat offspring after intrauterine ethanol exposure. Am J Physiol Regul Integr Comp Physiol. 2007;292(5):R1926–33.
Yao XH, Chen L, Nyomba BL. Adult rats prenatally exposed to ethanol have increased gluconeogenesis and impaired insulin response of hepatic gluconeogenic genes. J Appl Physiol. 2006;100(2):642–8.
Xia LP, Shen L, Kou H, Zhang BJ, Zhang L, Wu Y, et al. Prenatal ethanol exposure enhances the susceptibility to metabolic syndrome in offspring rats by HPA axis-associated neuroendocrine metabolic programming. Toxicol Lett. 2014;226(1):98–105. This study demonstrated that prenatal ethanol exposure induces increased susceptibility to adult metabolic syndrome in male rat offspring fed with high-fat diet.
Williams KH, Shackel NA, Gorrell MD, McLennan SV, Twigg SM. Diabetes and nonalcoholic fatty liver disease: a pathogenic duo. Endocr Rev. 2013;34(1):84–129.
Shen L, Liu Z, Gong J, Zhang L, Wang L, Magdalou J, et al. Prenatal ethanol exposure programs an increased susceptibility of non-alcoholic fatty liver disease in female adult offspring rats. Toxicol Appl Pharmacol. 2014;274(2):263–73.
Harper KM, Tunc-Ozcan E, Graf EN, Redei EE. Intergenerational effects of prenatal ethanol on glucose tolerance and insulin response. Physiol Genomics. 2014;46(5):159–68.
Dobson CC, Mongillo DL, Brien DC, Stepita R, Poklewska-Koziell M, Winterborn A, et al. Chronic prenatal ethanol exposure increases adiposity and disrupts pancreatic morphology in adult guinea pig offspring. Nutr Diabetes. 2012;2:e57.
Dobson CC, Thevasundaram K, Mongillo DL, Winterborn A, Holloway AC, Brien JF, et al. Chronic prenatal ethanol exposure alters expression of central and peripheral insulin signaling molecules in adult guinea pig offspring. Alcohol. 2014;48(7):687–93.
Bauldry S, Shanahan MJ, Boardman JD, Miech RA, Macmillan R. A life course model of self-rated health through adolescence and young adulthood. Soc Sci Med. 2012;75(7):1311–20.
SAMHSA (Substance Abuse and Mental Health Services Administration). Results from the 2010 national survey on drug use and health: summary of national findings. Office of Applied Studies, NSDUH Series H-41, Rockville, Md, USA, 2011.
Plessinger MA, Woods Jr JR. Cocaine in pregnancy. Recent data on maternal and fetal risks. Obstet Gynecol Clin N Am. 1998;25(1):99–118.
Cressman AM, Natekar A, Kim E, Koren G, Bozzo P. Cocaine abuse during pregnancy. J Obstet Gynaecol Can. 2014;36(7):628–31.
Lipshultz SE, Frassica JJ, Orav J. Cardiovascular abnormalities in infants prenatally exposed to cocaine. J Pediatr. 1991;118:44–51.
Messiah SE, Lipshultz SE, Miller TL, Accornero VH, Bandstra ES. Assessing latent effects of prenatal cocaine exposure on growth and risk of cardiometabolic disease in late adolescence: design and methods. Int J Pediatr. 2012;2012:467918.
Messiah SE, Miller TL, Lipshultz SE, Bandstra ES. Potential latent effects of prenatal cocaine exposure on growth and the risk of cardiovascular and metabolic disease in childhood. Prog Pediatr Cardiol. 2011;31(1):59–65.
Shankaran S, Bann CM, Bauer CR, Lester BM, Bada HS, Das A, et al. Prenatal cocaine exposure and BMI and blood pressure at 9 years of age. J Hypertens. 2010;28(6):1166–75.
LaGasse LL, Gaskins RB, Bada HS, Shankaran S, Liu J, Lester BM, et al. Prenatal cocaine exposure and childhood obesity at nine years. Neurotoxicol Teratol. 2011;33(2):188–97.
Ganapathy V, Prasad PD, Ganapathy ME, Leibach FH. Drugs of abuse and placental transport. Adv Drug Deliv Rev. 1999;38(1):99–110.
Suzuki K, Minei LJ, Johnson EE. Effect of nicotine upon uterine blood flow in the pregnant rhesus monkey. Am J Obstet Gynecol. 1980;136(8):1009–13.
Aliyu MH, Wilson RE, Zoorob R, Brown K, Alio AP, Clayton H, et al. Prenatal alcohol consumption and fetal growth restriction: potentiation effect by concomitant smoking. Nicotine Tob Res. 2009;11(1):36–43.
Hayatbakhsh MR, Flenady VJ, Gibbons KS, Kingsbury AM, Hurrion E, Mamun AA, et al. Birth outcomes associated with cannabis use before and during pregnancy. Pediatr Res. 2012;71(2):215–9.
Liu AJ, Sithamparanathan S, Jones MP, Cook CM, Nanan R. Growth restriction in pregnancies of opioid-dependent mothers. Arch Dis Child Fetal Neonatal Ed. 2010;95(4):F258–62.
Huber G, Seelbach-Göbel B. Substance abuse and pregnancy from an obstetric point of view. Z Geburtshilfe Neonatol. 2014;218(4):142–8.
Ross LJ, Wilson M, Banks M, Rezannah F, Daglish M. Prevalence of malnutrition and nutritional risk factors in patients undergoing alcohol and drug treatment. Nutrition. 2012;28(7-8):738–43.
Santolaria F, Gonzalez-Reimers E. Alcohol and nutrition: an integrated perspective. In: Watson RR, Preedy VR, editors. Nutrition and alcohol: linking nutrient interactions and dietary intake. Boca Raton: CRC Press; 2004. p. 3–17.
Zoli M, Picciotto MR. Nicotinic regulation of energy homeostasis. Nicotine Tob Res. 2012;14(11):1270–90.
Ersche KD, Stochl J, Woodward JM, Fletcher PC. The skinny on cocaine: insights into eating behavior and body weight in cocaine-dependent men. Appetite. 2013;71:75–80.
Aubin HJ, Farley A, Lycett D, Lahmek P, Aveyard P. Weight gain in smokers after quitting cigarettes: meta-analysis. BMJ. 2012;345:e4439.
Sofuoglu M, Dudish-Poulsen S, Poling J, Mooney M, Hatsukami DK. The effect of individual cocaine withdrawal symptoms on outcomes in cocaine users. Addict Behav. 2005;30(6):1125–34.
Cocores JA, Gold MS. The Salted Food Addiction Hypothesis may explain overeating and the obesity epidemic. Med Hypotheses. 2009;73(6):892–9.
Vik T, Jacobsen G, Vatten L, Bakketeig LS. Pre- and post-natal growth in children of women who smoked in pregnancy. Early Hum Dev. 1996;45:245–55.
Ross MG, Desai M. Developmental programming of offspring obesity, adipogenesis, and appetite. Clin Obstet Gynecol. 2013;56(3):529–36.
Hanson MA, Gluckman PD. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev. 2014;94(4):1027–76.
Acknowledgments
The author would like to thank Oksana Zabuga for the technical assistance in preparing this manuscript.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
A.M. Vaiserman declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Lifestyle Management to Reduce Diabetes/Cardiovascular Risk
Rights and permissions
About this article

Cite this article
Vaiserman, A.M. Early-Life Exposure to Substance Abuse and Risk of Type 2 Diabetes in Adulthood. Curr Diab Rep 15, 48 (2015). https://doi.org/10.1007/s11892-015-0624-3
Published:
DOI: https://doi.org/10.1007/s11892-015-0624-3