
Abstract
In addition to its central role in the development of microvascular complications, hyperglycemia plays an important role in the pathogenesis of type 2 diabetes mellitus (T2DM) by means of glucotoxicity. Thus, effective glycemic control not only reduces the incidence of microvascular complications but also corrects the metabolic abnormalities that contribute to the progression of the disease. Progressive β-cell failure and multiple side effects, including hypoglycemia and weight gain, associated with many current therapies present obstacles to the achievement of optimal and durable glycemic control in subjects with T2DM. Most recently, inhibitors of the renal sodium-glucose cotransporter have been developed to reduce the plasma glucose concentration by producing glucosuria. Because the mechanism of action of these oral antidiabetic agents is independent of β-cell function and tissue sensitivity to insulin, they improve glycemic control while avoiding hypoglycemia and promoting weight loss. In this review, we summarize the available data concerning the mechanism of action, efficacy, and safety of this novel antidiabetic class of therapeutic agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hyperglycemia is the major risk factor for microvascular complications of diabetes. Many studies have documented that every 1 % decrease in hemoglobin A1c (HbA1c) is associated with an approximately 35 % reduction in the risk of microvascular complications [1, 2]. Hyperglycemia also plays an important role in the pathogenesis of insulin resistance and β-cell failure (ie, glucotoxicity) [3, 4]. Thus, appropriate glycemic control in people with diabetes would be anticipated not only to reduce the risk of microvascular complications but also to ameliorate the metabolic abnormalities that contribute to the progressive course of the disease. Despite the irrefutable evidence for the importance of maintaining optimal glycemic control (HbA1c < 6.5 % to 7 %), about one half of people with diabetes in the United States fail to achieve the American Diabetes Association target for glycemic control and manifest HbA1c greater than 7 % [5, 6].
Progressive β-cell failure, weight gain, and hypoglycemia represent major obstacles to the achievement of tight glycemic control, HbA1c ≤ 6.5 % to 7 %, in patients with type 2 diabetes mellitus (T2DM) [3]. Therefore, the development of novel medications that effectively lower the plasma glucose concentration and produce durable glycemic control while avoiding hypoglycemia and weight gain are needed for the management of T2DM patients. Most recently, inhibitors of the renal sodium-glucose cotransporter (SGLT) have been developed to reduce the plasma glucose concentration by producing glucosuria [7••]. In this review, we summarize the available data concerning the mechanism of action, efficacy, and safety of this novel antidiabetes therapeutic approach.
Filtration and Reabsorption of Glucose by the Kidney
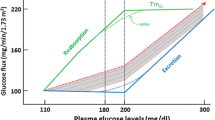
The kidney filters approximately 180 L of plasma each day, which contains about 162 g of glucose. In subjects with normal glucose tolerance virtually all of this glucose is reabsorbed in the proximal tubule. The maximum glucose transport capacity (Tm) of the proximal tubule, on average, has a value of approximately 375 mg/min [8]. Renal glucose reabsorption takes place in the proximal tubule primarily in the S1 and S3 segments (Fig. 1a). SGLTs, which couple glucose reabsorption to sodium reabsorption, mediate renal glucose reabsorption. The sodium electrochemical gradient generated by active sodium transport provides the energy required for glucose transport. SGLT2 has low affinity and high capacity for glucose transport. SGLT2 mediates glucose transport in the S1 segment and absorbs about 80 % to 90 % of filtered glucose. SGLT1 has high affinity but low capacity for glucose transport. It mediates glucose transport in the S3 segment and reabsorbs the remaining 10 % to 20 % of filtered glucose. In normal individuals, the filtered glucose load is less than 375 mg/min. Thus, all of the filtered glucose is reabsorbed and returned to the circulation. However, if the filtered glucose load exceeds 375 mg/min, as often occurs in subjects with poorly controlled T2DM, the Tm is exceeded and all of the glucose in excess of the Tm is excreted in the urine. The plasma glucose concentration at which the filtered glucose load reaches 375 mg/min is called the threshold (Fig. 1b). When the threshold is exceeded, the glucose excretion rate increases linearly and parallels the filtered load. The reabsorption and excretion curves display a nonlinear transition as the Tm for glucose is approached. This “rounding” of the curves is termed splay, and has been explained by heterogeneity in the Tm of individual nephrons and/or glomerulotubular imbalance [8].

a Renal handling of glucose. b Kinetics of renal glucose reabsorption. (Adapted with permission from: Abdul-Ghani MA, Norton L, Defronzo RA Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 32:515–31, 2011) [7••]
Subjects with normal glucose tolerance have a Tm for glucose that is well above the filtered glucose load. This has major survival benefits, since it allows the kidneys to conserve this critical energy source for the brain, which (with the exception of prolonged fasting) can utilize only glucose to generate energy for neuronal function. Individuals with T2DM have increased renal Tm for glucose [9, 10]. It has been postulated that the increase in Tm represents an adaptive mechanism to preserve energy during conditions when food was sparse. Today, where food is abundant and diabetes has reached epidemic proportions, this adaptive mechanism has become maladaptive. In patients with diabetes it would be desirable for the kidney to excrete the excess filtered glucose load and restore normoglycemia. In contrast, the increased Tm for glucose minimizes glucosuria and exacerbates the hyperglycemia.
It also should be noted that increased glucose uptake in the proximal tubule in subjects with diabetes is expected to be accompanied by increased sodium reabsorption. One can speculate that the increased sodium reabsorption can lead to extracellular volume expansion and an increase in blood pressure. Although this hypothesis has never been tested in humans, a recent micropuncture study in experimental animals has demonstrated that acute inhibition of SGLT2 causes a two- to threefold increase in single nephron sodium excretion [11]. However, the increased sodium excretion waned after 2 weeks, suggesting an important role of tubuloglomerular feedback in the long-term fluid and salt balance [11].
Based upon these pathophysiologic considerations, it follows that development of specific inhibitors of the renal SGLT2 transporter provides a rational and novel approach for the treatment of diabetic patients. This approach will, not only decrease plasma glucose concentration, but can be expected to have additional metabolic benefits such as lowering blood pressure and promoting weight loss. The specificity of drugs that inhibit SGLT2 over SGLT1 (which is present in both the gut and kidney) avoids impaired intestinal glucose absorption and diarrhea. Persistent glucosuria potentially could cause an increase in urinary tract infections and impair the kidney’s ability to concentrate urine. However, the fact that subjects with familial glucosuria are asymptomatic suggest that, if these side effects occur, they are likely to be mild.
Pharmacological Inhibitors of Renal Glucose Uptake
Phlorizin, isolated from the bark of apple trees, was the first SGLT inhibitor to be identified [12, 13]. It is comprised of two main moieties, a glucose ring connected via an oxygen atom (O-glucoside) to two phenol rings. It competitively inhibits both SGLT1 and SGLT2 in the proximal tubule with a higher affinity (10-fold) for the SGLT2 versus SGLT1 transporter. Intravenous injection of phlorizin in normal subjects produces glucosuria, resembling familial renal glucosuria [13]. Despite the efficacy of phlorizin in inhibiting SGLT2 transporter activity and normalizing the plasma glucose concentration in diabetic animals, low bioavailability (~ 15 %) following oral administration and inhibition of SGLT1 in the gastrointestinal tract negate its clinical usefulness in human subjects with diabetes [14].
Based upon the structure of phlorizin several other compounds with greater bioavailability following oral administration and higher selectivity for SGLT2 compared to SGLT1 have been developed (Table 1) and are in varying stages of development for clinical use. A second family of non-glucoside SGLT2 inhibitors with even greater selectivity for SGLT2 has been identified [15], but none of the members of this novel group of SGLT2 inhibitors is currently under clinical development.
Downregulation of SGLT2 gene expression with antisense oligonucleotides (ASOs) represents another approach that has been used to inhibit renal glucose reabsorption [16]. Studies in rats, dogs, and monkeys have demonstrated that ASOs decrease renal SGLT2 mRNA expression by about 80 % with no significant change in SGLT1 expression. This is accompanied by pronounced glucosuria. Further, a once-weekly injection of ASO for 4 to 5 weeks caused a substantial reduction in plasma glucose concentration and HbA1c in these diabetic animal models without side effects. Because the ASOs work by reducing the SGLT2 protein content, rather than inhibiting the SGLT2 transporter, they have the potential to cause a much greater reduction in HbA1c.
Inhibition of Renal Glucose Transport Corrects Hyperglycemia: Proof of Concept
Studies performed with phlorizin in 90 % pancreatectomized diabetic rats have provided proof of concept for the efficacy of SGLT2 inhibition in the treatment hyperglycemia. Chronic phlorizin administration induced glucosuria and normalized both the fasting and fed plasma glucose levels. Phlorizin also completely reversed insulin resistance and corrected the defects in both first- and second-phase insulin secretion [17–19]. The results of these studies demonstrate that inducing glucosuria by inhibiting renal SGLT2 is a promising strategy to correct hyperglycemia in diabetic individuals.
Familial renal glucosuria, a benign condition, has provided assurance for the safety of pharmacological inhibition of SGLT2. Affected individuals with loss-of-function mutations in the gene encoding for the SGLT2 transporter manifest varying degrees of glucosuria (20–200 g/d). Despite the glucosuria, these subjects are asymptomatic and do not experience hypoglycemia [20]. These observations provide proof of concept that pharmacological inhibition of SGLT2 is a safe and potentially effective strategy for reducing the plasma glucose concentration in diabetic subjects.
SGLT2 Inhibitors in T2DM Subjects
O-Glucosides: The First SGLT2 Inhibitors
The early SGLT2 inhibitors with high specificity for SGLT2 over SGLT1 were derivatives of phlorizin. T-1095 was the first agent to be developed [21], but preclinical studies demonstrated that its selectivity for SGLT2 was not satisfactory and it was not developed further. Sergliflozin and remogliflozin, which have greater selectivity to SGLT2 than T-1095, were subsequently developed [22]. In a phase 1 study, a single dose of sergliflozin (50–500 mg) caused a dose-dependent increase in glucosuria in both normal and T2DM subjects [23, 24]. The 500-mg dose reduced the mean plasma glucose concentration during the oral glucose tolerance test (OGTT) from 18.3 to 11.2 mM [24]. More prolonged treatment (14 days) with sergliflozin also induced dose-dependent glucosuria. Interestingly, SGLT2 inhibition was accompanied by an increase in plasma glucagon-like peptide-1 concentration and weight loss of 1.5 kg [24]. Although remogliflozin differs in its structure from phlorizin, it also has an O-glucoside linkage [25]. Remogliflozin causes dose-dependent glucosuria. In 35 drug naïve T2DM subjects, 12 days of treatment resulted in decrease in fasting plasma glucose (FPG) concentration (~30 mg/dL), body weight (2.6 kg), and blood pressure (~8 mmHg) [26].
For unknown reasons (described by the sponsor as “evaluating circumstances”), the development of both sergliflozin and remogliflozin has been discontinued and they were replaced with other SGLT2 inhibitors [27].
C-glucoside Inhibitors
Substitution of the O-link with a C-link provides greater resistance to β-glucosidase and has resulted in the development of longer-acting SGLT2 inhibitors, which are appropriate for single daily dosing. Many members of this group currently are in varying stages of clinical development.
Dapagliflozin.
Clinical trials with dapagliflozin are the most advanced among the C-glucoside SGLT2 inhibitors. Phase 3 trials have been completed and reviewed by the US Food and Drug Administration (FDA) [28••]. Initial dose-ranging studies demonstrated that dapagliflozin (5, 25, 100 mg/d) produces glucosuria (37, 62, 80 g/24 h, respectively) and significantly decreases the FPG concentration (by 19, 29, and 39 mg/dL, respectively) and the incremental area under the glucose curve concentration during an OGTT over 14 days of treatment in subjects with T2DM [29]. In humans, dapagliflozin has a T ½ about 17 to 18 h, making it suitable for once-daily administration [30]. Dapagliflozin is rapidly absorbed after oral administration, achieving maximal plasma concentrations within 2 h. It does not inhibit or induce P450 enzymes. Dapagliflozin is highly protein bound (97 % to 98 %) and renal excretion is low (2 % to 4 %). An inert glucuronide conjugate (M15) of dapagliflozin is the major metabolite of the drug in vivo.
More prolonged treatment (12 weeks) with dapagliflozin reduced the HbA1c by about 0.7 % without any apparent dose dependency in 389 T2DM subjects with a baseline HbA1c of 7.8 % to 8.0 % [31]. The reduction in HbA1c was similar in magnitude to that observed with metformin, and the reductions in fasting and postprandial plasma glucose concentrations accounted approximately equally for the decline in HbA1c [31]. Dapagliflozin treatment promoted between 2.2- and 3.1-kg weight loss and a modest reduction in both systolic and diastolic blood pressure. The amount of glucosuria observed with dapagliflozin (50–60 g/d) is equivalent to a daily caloric loss of about 200 to 240 cal/day, which over the course of 12 weeks could explain the 2- to 3-kg weight loss.
In five phase 3 trials [28••], treatment with dapagliflozin (N = 6798 randomized to dapagliflozin and placebo in a 2:1 ratio; 5 and 10 mg/day) consistently caused a significant decrease in HbA1c (>0.5 %) compared to placebo, independent of the background therapy. A similar decrease in HbA1c was observed when dapagliflozin was given as monotherapy or added to metformin, sulfonylurea, thiazolidinedione, or insulin. The decrease in HbA1c could be explained by a decrease in both fasting and postprandial plasma glucose concentrations. The 10-mg/day dapagliflozin dose produced an approximate 25 and 55 mg/dL decrease in the fasting and 2-hour plasma glucose concentrations, respectively. The decrease in HbA1c caused by dapagliflozin was independent of gender, ethnicity, race, body mass index, or duration of T2DM. As expected, dapagliflozin produced a greater reduction in HbA1c in patients with higher baseline HbA1c. In a 24-week study, dapagliflozin reduced the HbA1c by 2.88 % (5 mg/d) and 2.66 % (10 mg/d) in a subgroup (n = 78) of subjects with baseline HbA1c 10.1 % to 12.0 % [32].
Because the mechanism of action of dapagliflozin is independent of insulin secretion and insulin action, the efficacy of dapagliflozin is independent of β-cell function or diabetes duration. In one study, Wilding et al. [33] randomized 71 insulin-treated (≥50 units/d) type 2 diabetic patients who also were receiving an insulin sensitizer (metformin and/or thiazolidinedione) to add-on therapy with dapagliflozin (5 and 10 mg/d) or placebo. The insulin dose was reduced by 50 % at the start of therapy, while the insulin sensitizer dose was unchanged. After 12 weeks, the placebo-subtracted declines in HbA1c were 0.70 % and 0.78 %, respectively (P < 0.01 vs placebo), despite the 50 % reduction in insulin dose. The placebo-subtracted reductions in body weight were 2.6 and 2.4 kg, respectively (P < 0.01 vs placebo), reflecting both the loss of glucose in the urine and reduction in insulin dose. In another study, 151 subjects with new-onset diabetes (<1 year) and 58 subjects with longstanding (11 years) type 2 diabetes were randomly assigned to 10 or 20 mg/day of dapagliflozin for 12 weeks [34]. Subjects with longstanding diabetes were in poor glycemic control (HbA1c 8.4 %), despite large doses of insulin (>50 units/d) plus metformin and a thiazolidinedione. Dapagliflozin caused a comparable decline in HbA1c in both groups.
In a head-to-head comparison of dapagliflozin with sulfonylurea as add-on therapy in poorly controlled T2DM individuals on metformin therapy [35], both groups exhibited the same decline in mean HbA1c (−0.52 %). However, it should be noted that the subjects in this study had a relatively low initial HbA1c, and 25 % of participants had baseline HbA1c less than 7 %.
The results of 2-year follow-up with dapagliflozin treatment have recently been reported in two trials [36, 37]. In one study in which dapagliflozin treatment was added to subjects treated with metformin, the decrease from baseline in HbA1c was −0.54 % at 1 year and −0.80 % at 2 years. Similarly, in a head-to head study of dapagliflozin versus glipizide, an additional −0.34 % decrease in HbA1c was observed in the second year in subjects treated with dapagliflozin compared to a −0.12 % decrease in subjects treated with glipizide [37]. About 70 % and 90 % of subjects in the two studies [36, 37], respectively, entered the second-year extension study. Thus, it is difficult to determine whether the decrease in HbA1c during the second year was due to dapagliflozin action or due to selection bias in the subjects entering the second-year extension study. Nevertheless, the results of both studies demonstrate that the glucose-lowering effect of dapagliflozin dose not wane with time up to a 2-year period.
Canagliflozin
In type 2 diabetic individuals, canagliflozin produced dose-dependent glucosuria with a maximal effect at 400 mg per day. The glucosuria was accompanied by a dose-dependent decrease in the incremental area under both the plasma glucose and insulin concentration curves during a mixed meal [38]. Canagliflozin was effective in lowering the plasma glucose concentration as monotherapy and as add-on to subjects treated with metformin and insulin. In a 12-week placebo-controlled trial, canagliflozin monotherapy resulted in a dose-dependent decrease in HbA1c with a maximal placebo-subtracted decrease in HbA1c of 0.99 % with a daily dose of 300 mg [39]. In a double-blind, placebo-controlled, dose-ranging study in 451 T2DM subjects treated with metformin, canagliflozin in doses 50, 100, 200, and 300 mg/day for 12 weeks reduced the HbA1c by 0.7 % to 0.9 % from baseline and by 0.5 % to 0.7 % versus placebo in association with weight loss of 1.3 to 2.3 kg [40]. In 29 diabetic subjects treated with insulin, the addition of canagliflozin (100 and 300 mg/d) caused a −0.54 % and −0.73 % placebo-subtracted decrease in HbA1c after 28 days of treatment [41]. The 300-mg/day dose appeared to be slightly more effective then the lower doses. In a 16-day trial canagliflozin was shown to improve β-cell function in type 2 diabetic patients using a model-based method to calculate insulin secretion [42].
Empagliflozin
Similar to other SGLT2 inhibitors, empagliflozin produced a dose-dependent glucosuria in both normal and T2DM subjects [43, 44]. In a 4-week study, empagliflozin (100 mg/d) caused urinary glucose excretion of 74 g per day [45]. In a placebo-controlled 12-week study in 495 diabetic subjects with poor glycemic control on metformin, empagliflozin caused a dose-dependent decrease in both the FPG concentration and HbA1c with a placebo-subtracted decrease in FPG and HbA1c of 27 mg/dL and 0.7 %, respectively, with a dose of 25 mg per day [46].
Ipragliflozin
In a 12-week double-blind study, 361 Japanese type 2 diabetic subjects treated with ipragliflozin at doses ranging from 12.5 to 100 mg/day experienced a 0.9 % reduction in HbA1c at the two highest doses (50 and 100 mg/d) [47]. Body weight also was dose-dependently reduced by up to 2 kg in the 100 mg/day dose. In a 16-week study, ipragliflozin monotherapy (50 mg/d) caused a 1.2 % decrease from baseline (8.3 %) in HbA1c in 62 type 2 diabetic individuals [48].
LX4211
In a phase 2A study, LX4211, which inhibits SGLT2 and to a lesser extent SGLT1, at doses of 150 and 300 mg/day reduced the HbA1c by 1.2 %, but the starting HbA1c (8.2 % to 8.5 %) was higher than in most other studies and the placebo decreased the HbA1c by 0.5 % [49].
TS-071
In a 12-week phase 2 placebo-controlled study, TS-071 caused a dose-dependent decrease in HbA1c in 236 Japanese T2DM patients. The placebo-corrected decrease in HbA1c was 0.82 with a 5-mg per day dose [50].
PF04971729
In a 12-week double-blind, placebo-controlled phase 2 study in 328 subjects with T2DM, PF04971729 caused a dose-dependent decrease in HbA1c. The placebo-subtracted decrease in HbA1c with the highest dose (25 mg/d) was 0.72 %, and it was comparable to the placebo-subtracted decrease in HbA1c caused by sitagliptin (0.76 %). The decrease in HbA1c produced by PF04971729 was accompanied with an approximately 30 mg/dL decrease in the FPG concentration compared to a 17 mg/dL decrease in the sitagliptin-treated group [51].
It is noteworthy that the increase in urine glucose excretion (60–80 g/d) observed with all SGLT2 inhibitors currently in clinical trials, even with maximal doses, represents inhibition of less than 50 % of the filtered glucose load. The failure to observe a greater inhibition of renal glucose absorption is unclear but could be explained by: 1) inability of the SGLT2 inhibitor to reach the SGLT2 transporters because of their anatomical location; 2) competitive inhibition that progressively raises the local concentration of glucose at the site of the SGLT2 transporter, thus reducing its effectiveness; 3) insufficiently high drug concentrations in the tubular lumen to inhibit the SGLT2 transporter; 4) glucose transporters other than SGLT2 may be responsible for a much greater fraction of glucose reabsorption than previously appreciated; and 5) upregulation of the SGLT1 or other glucose transporters offsets the glucosuric effect of SGLT2 inhibitors. The latter seems unlikely since the magnitude of glucosuria on days 1 to 3 versus day 14 after the start of dapagliflozin is similar [28••]. Competitive inhibition by increased glucose concentration at the site of SGLT2 also seems unlikely, since increasing the dose of dapagliflozin from 20 to 500 mg failed to further increase renal glucose excretion.
Because of their mechanism of action, the efficacy of SGLT2 inhibitors to reduce the plasma glucose concentration is highly dependent upon renal function. Thus, as the glomerular filtration rate (GFR) decreases, there is a progressive impairment in glucosuria and a progressive decrease in the glucose-lowering ability of the drug. In subjects with a mild decrease in renal function (GFR = 60–90 mL/min), the glucosuria produced by dapagliflozin [28••] was decreased by 40 % and the reduction in HbA1c was decreased by about 22 % (Fig. 2). Interestingly, among subjects with similarly impaired renal function, ipragliflozin was reported to produce comparable glucosuria to subjects with GFR greater than 90 mL/min [52]; however, the decrease in FPG concentration was decreased by one half (12.9 mg/dL compared to 24.5 mg/dL, respectively). In subjects with moderately impaired renal function (GFR = 30–59 mL/min), the glucosuria produced by both ipragliflozin and dapagliflozin was markedly reduced (by~80 %) and the decrease in FPG and HbA1c was clinically insignificant (4 mg/dL and −0.11 %, respectively).

Impact of reduced renal function on the glucose-lowering efficiency of dapagliflozin. GFR glomerular filtration rate; HbA 1c hemoglobin A1c. (Adapted from [28••])
Lastly, it has been suggested that the oral SGLT2 inhibitors cause glucosuria by decreasing the Tm for glucose and/or increasing the glucose splay. One study in rodents with sergliflozin indicates a reduction in Tm without change in the glucose splay [22]. We believe that neither of these two explanations (reduced Tm or increased splay) satisfactorily can explain the marked glucosuria induced by the SGLT2 inhibitors in normal glucose–tolerant individuals with an FPG of 80 to 90 mg/dL. Rather, we believe that the SGLT2 inhibitors markedly reduce (to <50 mg/dL) the threshold at which glucose is spilled into the urine. This does not exclude a concomitant reduction in the glucose Tm, consistent with the effect of sergliflozin on renal glucose excretion [22].
SGLT2 Inhibitors and Diabetic Nephropathy
Hyperglycemia is the principal risk factor for diabetic microvascular complications (retinopathy, nephropathy, and neuropathy), and improved glycemic control—no matter how achieved—would be expected to reduce the risk of microvascular complications in subjects with type 2 diabetes [1, 2]. Because of the important role of enhanced sodium-glucose reabsorption in the proximal tubule in the development of diabetic nephropathy [53, 54], the SGLT2 inhibitors might be expected to have an additional beneficial renoprotective action beyond its glucose-lowering effect [53, 54]. The increased filtered glucose load in diabetes results in increased glucose and sodium reabsorption by the SGLT2 transporter in the proximal tubule [55, 56], and decreased sodium delivery to the juxtaglomerular apparatus. This activates the renin-angiotensin system resulting in elevated intraglomerular pressure and increased GFR [57]. By inhibiting sodium transport in the proximal tubule and increasing sodium delivery to the juxtaglomerular apparatus, renin and angiotensin secretion are inhibited, leading to a reduction in the glomerular pressure and hyperfiltration. Consistent with this, hyperfiltration and increased kidney size can be reversed by 6 weeks of intensive insulin therapy that normalizes the plasma glucose concentration [58]. With regard to this, it is noteworthy that chronic T-1095 administration decreased HbA1c levels in diabetic mice and stopped the progression of diabetic nephropathy, with prevention of proteinuria and expansion of glomerular mesangial area [59].
Nonglycemic Benefits
In addition to the beneficial effects related to improved glycemic control and possible renoprotective effects, the SGLT2 inhibitors have a number of nonglycemic effects that make them desirable agents as monotherapy and combination treatment. Weight gain is a major problem with currently available antidiabetic medications including sulfonylureas, thiazolidinediones, and insulin. The urinary loss of 60 to 80 g of glucose per day equates to 240 to 320 cal/day or 2 to 3 lb/month if this caloric deficit is not offset by an increase in caloric intake. Consistent with this, weight loss was observed in diabetic subjects treated with SGLT2 inhibitors in all clinical studies. A consistent finding in all dapagliflozin studies has been a reduction in systolic/diastolic blood pressure of 4–5/2–3 mm Hg [31]. Although this has been attributed to the mild fluid/sodium deficit that occurs during the first several days of dapagliflozin treatment [29, 31], an alternative explanation is local inhibition of the renin-angiotensin system secondary to enhanced sodium delivery to the juxtaglomerular apparatus [53, 54]. In one study, the decrease in blood pressure produced by PF04971729 treatment was comparable to that observed with thiazide diuretics. Because uric acid and sodium reabsorption in the proximal tubule are coupled, it is not surprising that a decrease in serum uric acid concentration has been observed in diabetic patients treated with dapagliflozin [54].
Safety
The pharmacological properties of the SGLT2 inhibitors suggest that they should have a good safety profile. The side effects observed in clinical studies were similar in all members of this class of drugs.
Because of their high selectivity for the SGLT2 transporter, no inhibition of the SGLT1 transporter in the intestinal mucosa is anticipated, and gastrointestinal side effects have not been observed. Furthermore, because subjects with homozygous mutations in the SGLT2 gene are asymptomatic despite large amounts of glucosuria (>50–100 g/24 h), pharmacological inhibition of SGLT2 would not be expected to cause polyuria, nocturia, or volume contraction. These side effects have not been observed in clinical trials. When dapagliflozin has been administered to humans for up to 12 to 24 weeks, urine volume increased only modestly (200–400 mL/d) during the first 2 to 3 days after initiation of therapy. Excessive urine loss of sodium, potassium, and other electrolytes was not observed [31]. Consistent with mild volume contraction, a small rise in hematocrit (1–2 volume %) and plasma urea nitrogen to creatinine ratio have been observed. Plasma electrolyte concentrations did not change following dapagliflozin [29, 31]. As stated previously, modest reductions in both systolic and diastolic blood pressure have been observed in type 2 diabetic patients [31]. Lastly, because SGLT2 inhibitors have no effect on the glucose counterregulatory mechanisms, hypoglycemia is not anticipated, and in nondiabetic and diabetic animals and in individuals with T2DM, SGLT2 inhibitors have not been associated with hypoglycemia [29, 31]. In individuals with T2DM treated with dapagliflozin at dose of 5 (n = 1145) and 10 (n = 1193) mg per day, an approximate 50 % increase in the incidence of urinary tract infection has been observed [28••]. It remains to be determined whether the glucosuria caused by SGLT2 inhibitors promotes bacterial growth. In one study in which mid-stream urine was collected, treatment with SGLT2 did not increase the prevalence of urinary bacteruria [60]. The incidence of vulvovaginitis and balanitis also are increased approximately twofold with SGLT2 inhibitor therapy [28••, 31–33].
SGLT2 inhibitors have not been shown to have any deleterious effect on renal function as manifested by a rise in serum creatinine or development of albuminuria or tubular proteinuria in subjects with T2DM with normal GFR [28••]. In phase 3 studies of dapagliflozin, an increased incidence of bladder and breast cancer was observed. However, the total number of cases was small (10 for each cancer). This finding was surprising because neither breast nor bladder tissue express the SGLT2 transporter. In addition, rigorous 2-year carcinogenic studies in animals failed to demonstrate any preneoplastic or neoplastic activity. Because breast and especially bladder cancer take many years to develop, whereas the exposure to dapagliflozin was short (generally <1 year), the significance of the increased incidence of these two tumors remains to be determined. Moreover, there could have been detection bias for bladder cancer due to the frequent testing for urinary tract infections, which could have led to the discovery of microscopic hematuria. Nonetheless, because of this potential carcinogenic signal, the FDA has asked for additional safety data regarding cancer risk with dapagliflozin treatment before approval of the drug for clinical use.
Conclusions
Current data in experimental animals and humans indicate that inhibition of the SGLT2 transporter is an effective and novel strategy to reduce the plasma glucose concentration in type 2 diabetic subjects. Dapagliflozin—the most clinically advanced of the SLGT2 inhibitors—has demonstrated a good safety profile, modest weight loss, a decrease in blood pressure, and HbA1c reduction of about 0.7 % to 0.8 % with a starting HbA1c of about 8.0 %. Because the SGLT2 inhibitors have a mechanism of action that is independent of insulin secretion or the presence of insulin resistance, the efficacy of this class of drugs is not anticipated to decline with progressive β-cell failure or in the presence of severe insulin resistance, although it will decline with declining renal function below GFR of 60 to 90 mL/min. Furthermore, this class of drugs can likely be used in combination with all other antidiabetic medications with anticipated additive efficacy on glycemic control. The SGLT2 inhibitors also are effective as monotherapy in newly diagnosed diabetic patients. To the extent that glucotoxicity contributes to the demise in β-cell function in subjects with impaired glucose tolerance or impaired fasting glucose, these drugs may also prove useful in the treatment of “prediabetes.” Currently available data indicate that the SGLT2 inhibitors have a good safety profile. In addition, the asymptomatic clinical presentation of subjects with familial renal glucosuria, despite multiple generations of the disease, supports the likely long-term safety of pharmacological inhibition of the SGLT2 transporter.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–86.
UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352:837–53.
DeFronzo RA. Banting lecture: from the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–95.
Rossetti L, Giaccari A, DeFronzo RA. Glucose toxicity. Diabetes Care. 1990;13:610–30.
The American Diabetes Association. Standards of medical care in diabetes-2007. Diabetes Care. 2007;30(Supp I):S4–41.
Hoerger TJ, Segel JE, Gregg EW, Saaddine JB. Is glycemic control improving in U.S. adults? Diabetes Care. 2008;31:81–6.
•• Abdul-Ghani MA, Norton L, Defronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 2011;32:515–31. This is a recent comprehensive review that summarizes the molecular basis of renal glucose reuptake and the clinical and pathophysiological implications of SGLT2 inhibitors..
Valtin H. Tubular reabsorption. In renal function. Boston: Little, Brown and Company; 1983.
Kamran M, Peterson RG, Dominguez JH. Overexpression of GLUT2 gene in renal proximal tubules of diabetic Zucker rats. J Am Soc Nephrol. 1997;8:943–8.
Farber SJ, Berger EY, Earle DP. Effect of diabetes and insulin of the maximum capacity of the renal tubules to reabsorb glucose. J Clin Invest. 1951;30:125–9.
Thomson SC, Rieg T, Miracle C, Mansoury H, Whaley J, Vallon V, Singh P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol. 2012;302:R75–83.
Chassis H, Jolliffe N, Smith H. The action of phlorizin on the excretion of glucose, xylose, sucrose, creatinine, and urea by man. J Clin Invest. 1933;12:1083–9.
Vick HD, Deidrich DF. Reevaluation of renal tubular glucose transport inhibition by phlorizin analogs. Am J Physiol. 1973;224:552–7.
Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21:31–8.
Li A, Zhang J, Greenberg J, Lee T, Liu J. Discovery of non-glucoside SGLT2 inhibitors. Bioorg Med Chem Lett. 2011;21:2472–5.
Bhanot S, Murray SF, Booten SL, Chakravarty K, Zanardi T, Henry S, Watts LM, Wancewicz EV, Siwkows A. ISIS 388626, an SGLT2 antisense drug, causes robust and sustained glucosuria in multiple species and is safe and well-tolerated. Diabetes. 2009;58(suppl1):A328.
Kahn BB, DeFronzo RA, Cushman SW, Rossetti L. Normalization of blood glucose in diabetic rats with phlorizin treatment reverses insulin-resistant glucose transport in adipose cells without restoring glucose transporter gene expression. J Clin Invest. 1999;87:561–70.
Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J Clin Invest. 1987;79:1510–5.
Rossetti L, Shulman GI, Zawalich W, DeFronzo RA. Effect of chronic hyperglycemia on in vivo insulin secretion in partially pancreatectomized rats. J Clin Invest. 1987;80:1037–44.
Santer R, Kinner M, Lassen CL, Schneppenheim R, Eggert P, Bald M, Brodehl J, Daschner M, Ehrich JH, Kemper M, Li Volti S, Neuhaus T, Skovby F, Swift PG, Schaub J, Klaerke D. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol. 2003;14:2873–82.
Oku A, Ueta K, Nawano M, Arakawa K, Kano-Ishihara T, Matsumoto M, Saito A, Tsujihara K, Anai M, Asano T. Antidiabetic effect of T-1095, an inhibitor of Na(+)-glucose cotransporter, in neonatally streptozotocin-treated rats. Eur J Pharmacol. 2000;391:183–92.
Katsuno K, Fujimori Y, Takemura Y, Hiratochi M, Itoh F, Komatsu Y, Fujikura H, Isaji M. Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J Pharmacol Exp Ther. 2007;320:323–30.
Hussey E, Clark R, Amin D, Kipnes M, Semmes R, Odriscoll E, Leong J, Murphy S, Dobbins R, Nunez D. Early clinical studies to assess safety, tolerability, pharmacokinetics and pharmacodynamics of single dose of sergliflozin, a novel inhibitor of renal glucose reabsorption in healthy volunteers and subjects with type 2 diabetes mellitus. Diabetes. 2007;56(Suppl1):A189.
Hussey E, Dobbins R, Stolz R, Stockman N, Semmes R, Murray S, Nunez D. A double-blind randomized repeat dose study to assess safety, tolerability, pharmacokineticks and pharmacodynamics of three times daily dosing of sergliflozin, a novel inhibitor of renal glucose reabsorption in healthy overweight and obese subjects. Diabetes. 2007;56(Suppl1):A491.
Fujimori Y, Katsuno K, Nakashima I, Ishikawa-Takemura Y, Fujikura H, Isaji M. Remogliflozin etabonate, in a novel category of selective low-affinity sodium glucose cotransporter (SGLT2) inhibitors, exhibits antidiabetic efficacy in rodent models. J Pharmacol Exp Ther. 2008;327:268–76.
Dobbins RL, Kapur A, Kapitza C, O’connor-Semmes RL, Tao W, Hussey EK. Remogliflozin etabonate, a selective inhibitor of the sodium-glucose transporter 2 (SGLT2) reduces serum glucose in type 2 diabetes mellitus (T2DM) patients. Diabetes. 2009;58 suppl 1:A573.
http://www.pharmaceutical-business-review.com/news/glaxosmithkline_discontinues_ development_of_remogliflozin_090703.
•• http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM262996.pdf This site contains the entire package for the FDA submission of dapagliflozin with a summary of the clinical efficacy and adverse events observed in phase 1, 2, and 3 of studies.
Komoroski B, Vachharajani N, Feng Y, Li L, Kornhauser D, Pfister M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharm Ther. 2009;85:513.
Obermeier M, Yao M, Khanna A, Koplowitz B, Zhu M, Li W, Komoroski B, Kasichayanula S, Discenza L, Washburn W, Meng W, Ellsworth BA, Whaley JM, Humphreys WG. In vitro characterization and pharmacokinetics of dapagliflozin (BMS-512148), a potent sodium-glucose cotransporter type II inhibitor, in animals and humans. Am Soc Pharmacol Exp Therapeut. 2010;38:405–14.
List J, Woo V, Morales E, Tang W, Fiedorek FT. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32:650–7.
Ferrannini E, Ramos SJ, Tang W, Salsali A, List JF. Dapagliflozin monotherapy in T2DM patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, multicenter phase III trial. Diabetes Care. 2010;33:2217–24.
Wilding JPH, Norwood P, Tjoen C, Bastien A, List JF, Fiedorek FT. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers. Applicability of a novel insulin-independent treatment. Diabetes Care. 2009;32:1656–62.
Zhang L, Feng Y, List J, Kasichayanula S, Pfister M. Dapagliflozin treatment in patients with different stages of type 2 diabetes mellitus: effects on glycaemic control and body weight. Diabetes Obes Metabol. 2010;12:510–6.
Nauck MA, Del Prato S, Meier JJ, Durán-García S, Rohwedder K, Elze M, Parikh SJ. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: a randomized, 52-week, double-blind, active-controlled noninferiority trial. Diabetes Care. 2011;34:2015–22.
Baily CJ, Gross JI, Yadav M, Iqbal N, Mansfield TA, List JF. Sustained efficacy of dapagliflozin when added to metformin in type 2 diabetes inadequately controlled by metformin monotherapy. Diabetologia. 2011;54(Suppl1):A146.
Del Prato S, Nauck M, Rohwedder K, Theuerkauf A, Langkilde AM, Parikh S. Long term efficacy and safety of ass-on dapagliflozin vs add-on glipizide in patients with type 2 diabetes mellitus inadequately controlled with metformin: 2 year results. Diabetologia. 2011;54(Suppl1):A852.
Sha S, Devineni D, Ghosh A, Polidori D, Chien S, Wexler D, Shalayda K, Demarest K. Rothenberg PCanagliflozin, a novel inhibitor of sodium glucose co-transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab. 2011;13:669–72.
Inagaki N, Kondo K, Iwasaki T, Maruyama N, Susuta Y, Sakai M, Kuki H. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2 (SGLT2) improves glycemic control and reduces body weight in Japanese type 2 diabetes Mellitus (T2DM). Diabetes. 2011;60(suppl):A999.
Rosensotck J, Arbit D, Usiskin K, Capuano G, Canovatchel W. Canagliflozin an inhibitor of sodium glucose co-transporter 2 (SGLT2), improves glycemic control and lowers body weight in subjects with type 2 diabetes (T2D) on metformin. Diabetes. 2010;59 suppl 1:A21.
Devineni D, Morrow L, Hompesch M, Skee D, Vandebosch A, Murphy J, Ways K, Schwartz S. Canagliflozin improves glycemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes Metab. 2012[ahead of print].
Polidori D, Zhao Y, Sha S, Canovatchel W. Canagliflozin treatment improves beta cell function in subject with type 2 diabetes. Diabetes. 2010;59 suppl 1:A176.
Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14:83–90.
Koiwai K, Seman L, Yamamura N, Macha S, Taniguchi A, Negishi T, Sesoko S, Dugi KA. Safety, tolerability, pharmacokinetics and pharmacodynamics of single doses of BI 10773, a sodium-glucose co-transporter inhibitor (SGLT2), in Japanese healthy volunteers. Diabetes. 2010;59 suppl 1:2175PO.
Heise T, Seewaldt-Becker E, Macha S, Hantel S, Huber K, Pinnetti S, Seman L, Hans-Juergen W. BI 10773, a sodium-glucose co-transporter inhibitor (SGLT-2), is safe and efficacious following 4-week treatment in patients with type 2 diabetes. Diabetes. 2010;59(suppl):629P.
Rosenstock J, Jelaska A, Seman L, Pinnetti S, Hantel S, Woerle HJ. Efficacy and safety of BI 10773, a new sodium glucose cotransporter-2 (SGLT-2) inhibitor, in type 2 diabetes inadequately controlled on metformin. Diabetes. 2011;60(suppl):989P.
Kashiwagi A, Utsuno A, Kazuta K, Yoshida S, Kageyama S. ASP1941, a novel, selective SGLT2 inhibitor, was effective and safe in Japanese healthy volunteers and patients with type 2 diabetes mellitus. Diabetes. 2010;59 suppl 1:A21.
Takinami A, Takinami Y, Kazuta K, Yoshida S, Utsuno A, Nagase I, Kashiwagi A. Ipragliflozin improved glycemic control with additional benefit of reduction of body weight and blood pressure in Japanese patients with type 2 diabetes mellitus BRIGHTEN Study. Diabetologia. 2011;54(suppl):A149.
Freiman J, Ruff DA, Frazier KS, Combs K, Turnage A, Shadoan M, Powell D, Zambrowicz B, Brown P. LX4211, a dual SGLT2/SGLT1 inhibtor, shows rapid and significant improvements in glycemic control over 28 days in patients with type 2 diabetes (T2DM). Diabetes 2010;59(suppl 1), (late breaking abstract).
Seino Y, Sasaki T, Fukatsu A, Samukawa Y, Sakai S, Watanabe T. TS-071, a novel and selective SGLT2 inhibitor, improved glycemic control and decreased body weight in 12-week study of Japanese patients with type 2 diabetes mellitus. Diabetes. 2011;60(suppl):P998.
Nucci G, Amin NB, Wang X, Lee DS, Rusnak JM. The sodium glucose co-transported PF04971729, provides multifaced improvement in diabetic patients inadequately controlled on metformin. Diabetologia. 2011;54(suppl):A850.
Kadokura T, Ishikawa H, Nakajo I, Yoshida S, Utsuno A, Smulders RA, Morozumi K. The effect of renal impairment on the pharmacokinetics and urinary glucose excretion of the SGLT2 inhibitor ipragliflozin in Japanese type 2 diabetes mellitus patients. Diabetologia. 2011;54(suppl):A847.
Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol. 1999;10:2569–76.
Thomson SC, Vallon V, Blantz RC. Kidney function in early diabetes: the tubular hypothesis of glomerular filtration. Am J Physiol. 2004;286:F8–18.
Noonan WT, Shapiro VM, Banks RO. Renal glucose reabsorption during hypertonic glucose infusion in female streptozotocin-induced diabetic rats. Life Sci. 2001;68:2967–77.
Dominguez JH, Camp K, Maianu L, Feister H, Garvey WT. Molecular adaptations of GLUT1 and GLUT2 in renal proximal tubules of diabetic rats. Am J Physiol. 1994;266:F283–90.
Nelson RG, Bennett PH, Beck GJ, Tan M, Knowler WC, Mitch WE, Hirschman GH, Myers BD. Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. N Engl J Med. 1996;335:1636–42.
Tuttle KR, Bruton JL, Perusek MC, Lancaster JL, Kopp DT, DeFronzo RA. Effect of strict glycemic control on renal hemodynamic response to amino acids and renal enlargement in insulin-dependent diabetes mellitus. N Engl J Med. 1991;324:1626–32.
Arakawa K, Ishihara T, Oku A, Nawano M, Ueta K, Kitamura K, Matsumoto M, Saito A. Improved diabetic syndrome in C57BL/KsJ-db/db mice by oral administration of the Na(+)-glucose cotransporter inhibitor T-1095. Br J Pharmacol. 2001;132:578–86.
Parikh S, Johnsson K, Ptaszynska A, Schmitz B, Sugg J, List JF. Characterization of urinary tract infection in the setting of pharmacologically induced glucosuria. Diabetologia. 2011;54(suppl):A841.
Disclosure
Conflicts of interest: M.A. Abdul-Ghani:none; L. Norton:none; R.A. DeFronzo: has board membership with Amylin, Takeda, Boehringer Ingelheim, Novo Nordisk, and Bristol-Myers Squibb; has received grant support from Amylin and Takeda, is a member of the Novo Nordisk speakers bureau.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abdul-Ghani, M.A., Norton, L. & DeFronzo, R.A. Efficacy and Safety of SGLT2 Inhibitors in the Treatment of Type 2 Diabetes Mellitus. Curr Diab Rep 12, 230–238 (2012). https://doi.org/10.1007/s11892-012-0275-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11892-012-0275-6