
Abstract
Myocardial infarction remains the commonest cause of premature death worldwide with coronary atherosclerotic plaque rupture often initiating the event. Despite an ever-expanding repertoire of cardiovascular imaging techniques, the race is still on to identify atherosclerotic lesions at high-risk of rupture: the so-called vulnerable plaque. Conventional imaging modalities such as stress testing and coronary angiography have consistently failed to identify such plaques, leading to the increasing appreciation that plaque rupture relates to factors other than just the degree of luminal stenosis. Indeed the focus has recently shifted to molecular imaging, in an attempt to directly target the pathological disease processes leading to rupture and thereby localize high-risk lesions. Histological data indicate that inflammation, necrosis and early stage microcalcification are key imaging targets by which to achieve this aim. Here, we discuss how these processes are related, focusing on the rationale and evidence supporting 18F-fluoride positron emission tomography as a novel non-invasive imaging technique for the identification of vulnerable atherosclerotic plaque.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases remain the commonest cause of death worldwide. The majority of these deaths are attributed to sudden atherosclerotic plaque rupture resulting in myocardial infarction or stroke [1]. However prediction of cardiovascular events is difficult because most are caused by atherosclerotic plaques that are non-flow limiting and therefore missed by conventional diagnostic modalities such as myocardial stress testing or invasive coronary arteriography. However, these high-risk plaques, the so-called vulnerable plaque, do have certain histopathological characteristics, which potentially can be targeted using modern imaging technology. Indeed the race is now on to identify these high-risk plaques in vivo using both invasive and non-invasive modalities [2, 3••, 4].
The Vulnerable Plaque
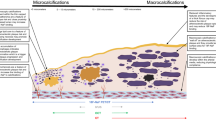
The concept of the vulnerable plaque was first introduced by James Muller in 1989 when he described ‘hemodynamically insignificant, albeit dangerous lesions’ [5], that he believed were at high risk of rupturing and causing myocardial infarction. Since then multiple observational studies have confirmed that most of the plaques causing myocardial infarction are non-flow limiting at the time of antecedent coronary arteriography. However pioneering work over the last two decades has now established that ruptured plaques do have certain key histopathological features including: the presence of a large necrotic core, a thin fibrous cap (<65 μm), a positively remodelled vessel, macrophage infiltration resulting in plaque inflammation, hypoxia leading to neovascularization and finally early stage microcalcification (Fig. 1) [6–11]. Moreover it is widely postulated that intact high-risk plaques are likely to demonstrate the same pathophysiological features immediately before a clinical plaque rupture event, so that identification of these characteristics may be of use prospectively. Recent technological advances, coupled with the failure of percutaneous coronary intervention to reduce myocardial infarction in patients with stable angina [12] has renewed interest in detecting these plaques in vivo. Indeed, this has been described by many as the ‘holy grail’ of clinical cardiology [1, 2, 3••, 13].

The relationship between inflammation, biphasic calcific response and adverse cardiovascular events. a Typical features of a vulnerable plaque: The initial stages of plaque inflammation and vulnerability are associated with macrophage influx into a large lipid core. By this stage, other features of plaque vulnerability such as positive remodelling, thinning of the fibrous cap, are observed. b Microcalcification within the necrotic core: Cell death occurring within the lipid core as a consequence of necrosis and apoptosis triggers microcalcification. This is a high risk plaque type that is ripe to rupture. This can have two consequences: successful plaque calcification by walling off the inflamed area, or initiation of plaque rupture with subsequent thrombotic occlusion. This is the plaque type that is believed to be have avid 18F-fluoride uptake on PET/CT. c Plaque rupture with thrombotic occlusion of the lumen resulting in myocardial infarction. d Plaque stabilization with successful healing of the necrotic core with obvious macrocalcification that can be detected with conventional imaging modalities such as computed tomography and X-ray angiography
The Link Between Inflammation and Calcification
Inflammation plays a critical role in the formation, progression, and rupture of atherosclerotic plaques and is typically characterized by the presence of macrophages within the plaque lipid core (Fig. 1) [6–11]. In an attempt to clear lipid from the vessel intima, these macrophages set up an inflammatory cycle that ultimately proves difficult to contain, leading to progressive matrix degradation and plaque destabilization. Indeed ongoing macrophage infiltration and cell death along with accelerated lipid accumulation contribute to an enlarging necrotic core that becomes progressively more inflamed and hypoxic. Moreover these cells secrete pro-inflammatory cytokines (including interleukin-1, monocyte chemotactic protein-1 and tumour necrosis factor-alpha) and matrix metalloproteinases, which actively weaken the fibrous cap: the only barrier between this highly thrombogenic lipid core and the vessel lumen. Fortunately this is where body defence mechanisms are believed to respond, triggering a calcific healing response that attempts to subdue and wall off this inflamed necrotic environment, thereby reducing the risk of plaque rupture.
Calcification occurs widely in the body and frequently occurring as a healing response to intense necrotic inflammation. This is perhaps best exemplified by tuberculosis where the body attempts to wall off the intense necrotic inflammation associated with caseating granulomata using calcification. Similar mechanisms are believed to occur within coronary atheroma, with calcification occurring as a healing response to intense inflammation within the necrotic core. However in these arteries, calcification appears to have a bi-phasic response, with each stage associated with markedly different plaque characteristics and clinical consequences. The latter phase of macroscopic calcification is readily imaged using standard x-ray angiography and CT, and is widely believed to impart stability to the plaque. Indeed by this stage the healing process has often successfully subdued inflammation within the vessel wall and separated it from the contents of the vascular lumen. By contrast the earlier phase of micro-calcification is not visible using standard non-invasive imaging techniques and is associated with plaque instability and an increased risk of rupture (Table 1) [14, 15]. The likely explanation is that at this early time-point the healing process has not yet been effective and that the inflamed necrotic environment triggering calcification still exists within the plaque. This risk of plaque rupture related to inflammation therefore persists. However recent data have indicated that in addition micro-calcification might itself increase the propensity to rupture, acting as a focal point that intensifies mechanical stresses on the surface of the cap. Either way a technique capable of directly imaging active ongoing micro-calcification and differentiating it from dormant macroscopic areas would hold real promise as a means of improving our understanding of plaque biology and in identifying high-risk atheroma.
Although there have been significant advances in our understanding as to why atherosclerotic plaques calcify, the exact molecular mechanisms underpinning this observation remain unclear. Multiple potential pathways have been proposed although often these have been established using models of vascular medial calcification. Calcification in the coronary arteries is almost uniquely intimal with medial and adventitial involvement occurring only rarely in conditions such as renal failure. Calcification activity in the intima appears to be closely related to inflammation and cell death within the necrotic core. Several putative pathways have been proposed linking these entities [16–21]. First and most obviously, necrosis of foam cells, vascular smooth muscle cells and other cells in the inflammatory plaque milieu leads to the release of large quantities of phosphate and calcium which may lead to spontaneous dystrophic calcification (as observed in infarctions, haematomas and scars). Second, inflammation may trigger osteogenic metaplasia in a variety of cell types (VSMC, endothelial cells, etc.) which make a phenotypic switch to osteoblast like cells under the influence of RUNX-2 transcription and become capable of roughly recapitulating skeletal osteogenesis in the plaque matrix. Third, circulating osteoprogenitors may be recruited to the plaque before undergoing maturation to classical osteoblasts. Fourth, cells undergoing programmed cell death within the plaque may provide calcifying substrate through the release of apoptotic bodies. Fifth and finally, macrophages themselves may provide the substrate for calcification by directly releasing matrix vesicles (the key and final executors of ordered tissue mineralization) into the extracellular matrix. Aikawa et al. have published the key and highly elegant longitudinal experiments in a mouse model that have conclusively demonstrated the link between inflammation and calcification [14]. They showed that macrophage infiltration is closely associated with osteogenic activity (as assayed by accumulation of OsteoSense; a bisphosphonate-conjugated to a fluorescent reporter). They (and others) have also shown that apoptotic bodies and matrix vesicles that contain calcium orthophosphate nanocrystals execute this early calcific process [20, 22, 23].
Positron Emission Tomography
Combined positron emission and computed tomography (PET/CT) is a modern non-invasive imaging technique that combines functional information from PET with the fine anatomical detail provided by CT, allowing the activity of specific pathological processes to be studied within even small structures in the body. This technique has been widely used in the clinical assessment of patients with cancer for many years, resulting in the widespread availability of PET/CT scanners [24]. Recent technological advances including ECG-gating, improved PET resolution and fusion with detailed CT angiography of the coronary vessels, has allowed translation of this technology into the heart. Theoretically any pathological process can be studied dependent on a suitable radiotracer being developed, so that potentially each of the established characteristics associated with high-risk plaque may be targeted. However to date the majority of studies investigating coronary atherosclerosis have utilized the tracers 18F-fluorodeoxyglucose (18F-FDG) and 18F-fluoride as markers of inflammation and micro-calcification respectively. Whilst a comprehensive discussion of 18F-FDG activity in the vasculature is beyond the scope of this review, it has become clear that whilst an excellent tracer for imaging vascular inflammation and perhaps hypoxic inflammation in the aorta and carotid arteries, utilization of this tracer in the coronary arteries is problematic. In particular, difficulty has arisen from uptake of this tracer by the adjacent myocardium, which frequently obscures the coronary signal. Whilst ultimately this problem may be rectified using advanced motion correction algorithms and attempts to switch myocardial metabolism away from glucose, further work is required before 18F-FDG is likely to prove useful in the coronary circulation. By contrast 18F-fluoride demonstrates an excellent signal to noise ratio in the coronary arteries as it is not taken up by adjacent structures and is rapidly cleared from plasma moreover it already appears capable of providing important clinical information with respect to coronary plaque biology.
Mechanism of 18F-Fluoride PET Activity
18F-Fluoride is a PET tracer with favourable pharmacokinetic properties, first introduced by Blau and coworkers in 1962 for the study of bone disease [25–27]. After an intravenous injection, approximately 70 % of 18F-fluoride is plasma based with the remaining 30 % found in erythrocytes. Because of its small size and negligible protein binding, 18F-fluoride demonstrates almost complete clearance from the blood stream on first pass [28–30], resulting in low blood-pool activity. This coupled with its specificity for bone and vascular calcification ensures that it provides excellent signal to noise in these tissues with little contamination from adjacent structures.
The mechanism of 18F-fluoride uptake in bone is well established. First it diffuses via the capillary network into the bone extracellular fluid. Then it exchanges with hydroxyl groups on exposed regions of hydroxyapatite crystal on the bone surface, forming fluoroapatite. The intensity of the signal depends both on the bone blood flow but also upon the surface area of exposed hydroxyapatite, which is increased in regions of new bone formation and remodelling [25–27]. As a consequence 18F-fluoride has been extensively utilized as a marker of bone turnover and used to study various bone related clinical conditions such as Paget’s disease [31, 32], osteoporosis [33, 34], renal osteodystrophy [14], fracture healing [35], and osteonecrosis [36]. Moreover, 18F-fluoride PET has become widely established as the most sensitive imaging modality for the detection of malignant bone involvement, leading to its widespread and commercial availability [37–41].
We believe that very similar mechanisms underlie the uptake of 18F-fluoride in the vasculature. Given the nature of the tissue, blood supply is not likely to be a major factor. However like bone, hydroxyapatite is also the key structural component of vascular calcium, so that arterial 18F-fluoride uptake is likely to relate closely to the available surface area of this crystal. Transmission electron microscopy studies have demonstrated that during the early stages of calcification hydroxyapatite crystals are nanosized, very thin and long [42]. This results in a much larger surface area of hydroxyapatite for 18F-fluoride binding in the early stages of microcalcification compared to macroscopic calcification in which much of the hydroxyapatite is internalized and not accessible to the tracer. The hypothesis that 18F-fluoride preferentially binds to vascular micro-calcification activity has been strongly supported by some early yet detailed pre-clinical work performed by our collaborators at Cambridge University and will no doubt be the subject of intensive future study [43].
Clinical Studies Examining Vascular 18F-Fluoride Activity
Derlin et al. first described the vascular uptake of 18F-fluoride in 2010 in a retrospective analysis of patients with malignancy [44]. The authors noted increased 18F-fluoride uptake in large vessels such as the aorta, carotids and femoral vessels in about three quarters of the patients studied. Interestingly, only a fifth of all calcified plaques on CT demonstrated increased 18F-fluoride uptake, highlighting even at this early stage that 18F-fluoride provides different information to the presence of calcium on CT. Subsequent work by the same group retrospectively compared the distribution of 18F-fluoride and 18F-FDG uptake in oncology patients [45] and suggested that 18F-fluoride signal in the femoral vessels correlated with the calcified plaque burden and cardiovascular risk factors [46]. Beheshti and colleagues first described 18F-fluoride activity localizing to the heart [47] whilst we and other groups have confirmed increased 18F-fluoride activity in the aorta and importantly in the valves of patients with aortic stenosis [48••, 49], where it acts as a marker of calcification activity and predicts disease progression [50].
Coronary 18F-NaF PET Identifies High Risk Patients
We first described 18F-fluoride uptake in the coronary arteries as a novel marker of plaque biology in subjects with and without aortic valve disease [51••]. This demonstrated the feasibility and excellent reproducibility of this tracer in the coronary vasculature. Moreover increased uptake of this tracer localized to individual coronary plaques and importantly identified patients at increased cardiovascular risk, with those subjects having increased Framingham risk scores and prior MACE event rates. This study also confirmed that, as in the aorta, 18F-fluoride provided different information to the presence of coronary calcium on CT. Indeed we observed that >40 % of patients with coronary artery calcium scores >1000 Agatston Units did not have 18F-fluoride uptake, suggesting the ability of 18F- fluoride to distinguish between dormant pacified calcific disease and metabolically active ongoing micro-calcification.
Coronary 18F-Fluoride PET as a Marker of High-Risk Atherosclerotic Plaque
Perhaps the key incidental observation from our initial study came from a patient with aortic stenosis admitted with an inferior non-ST elevation myocardial infarction [51••]. Intense coronary 18F-fluoride uptake was localized to the exact site of the culprit lesion in this patient, despite them having severe three vessel disease and widespread coronary calcium on CT. Based on this observation and the wider results of this preliminary study, we designed a subsequent trial to investigate 18F-fluoride activity in patients with stable and unstable coronary artery disease [52••]. Our hypothesis was that 18F-fluoride would identify high-risk vulnerable plaques in patients with stable angina and ruptured plaques in patients with myocardial infarction.
We examined 40 patients with stable angina referred for invasive angiography, who all underwent 18F-NaF PET/CT imaging alongside CT coronary angiography, invasive coronary angiography and intravascular ultrasound. Increased 18F-fluoride activity localized to individual coronary plaques in ~40 % of patients. These plaques had multiple high-risk features on intravascular ultrasound and CT including micro-calcification, positive remodelling and a large necrotic core. It was not possible to undertake histological analysis of the 18F-fluoride signal in these patients. However instead we demonstrated that increased 18F-fluoride activity colocalized with histological evidence of increased macrophage accumulation, cell death and calcification activity in carotid endarterectomy specimens. These data would indicate that 18F-fluoride identifies high-risk atherosclerotic plaque. However the real question is whether these plaques go on to cause myocardial infarction. Ultimately this will require prospective studies but we examined this issue retrospectively, investigating 18F-fluoride uptake in 40 patients who had recently sustained a type 1 myocardial infarction. We observed increased 18F-fluoride uptake at the site of the culprit plaque in 93 % of these patients. Indeed activity was around 30 % higher in the culprit plaque than the maximum activity recorded anywhere else in the coronary vasculature and was independent of coronary artery stenting.
Conclusions and Future Directions
In combination we believe that the clinical studies to date have demonstrated that 18F-fluoride provides complementary information to the presence of calcium on CT and that this technique is able to identify high-risk and ruptured coronary atherosclerotic plaques. It therefore holds major promise as a method of identifying vulnerable plaque and in improving upon current methods for the prediction of myocardial infarction. Indeed if confirmed then this approach may have a significant role to play in our future approach to the treatment of coronary artery disease.
Two major questions still need to be addressed before the potential of 18F-fluoride can be realized. First the mechanism for 18F-fluoride uptake in the vasculatures needs to be confirmed over and above the work we have already performed in the aortic valve and carotid arteries. Second the ability of 18F-fluoride to predict myocardial infarction needs to be addressed in the context of a prospective observational study, ideally in a high-risk population.
We believe that it is improbable that all coronary plaques with increased 18F-fluoride activity will go on to cause a myocardial infarction. Indeed calcification is often likely to prove successful in stabilizing the plaque and even when rupture does occur this will be sub-clinical in the majority of cases (Fig. 1). As a consequence individual plaque-directed treatment strategies are also unlikely to be effective. Instead we believe that the future role of 18F-fluoride will be in identifying the vulnerable patient. Those subjects with evidence of metabolically active coronary atheroma who are at an increased risk of myocardial infarction and would therefore benefit from some of the newer medical therapies that appear to have profound effects on coronary plaque biology but do so at great expense.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part I. Circulation. 2003;108(14):1664–72.
Calvert PA, Obaid DR, O'Sullivan M, Shapiro LM, McNab D, Densem CG, et al. Association between IVUS findings and adverse outcomes in patients with coronary artery disease: the VIVA (VH-IVUS in Vulnerable Atherosclerosis) study. JACC Cardiovasc Imaging. 2011;4(8):894–901.
Stone GW, Maehara A, Lansky AJ, de Bruyne B, Cristea E, Mintz GS, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364(3):226–35. This landmark study looked at the natural history of coronary atherosclerotic plaques using radiofrequency intravascular ultrasound and provided key pathological insights into our understanding of coronary disease.
Rogers IS, Nasir K, Figueroa AL, Cury RC, Hoffmann U, Vermylen DA, et al. Feasibility of FDG imaging of the coronary arteries: comparison between acute coronary syndrome and stable angina. JACC Cardiovasc Imaging. 2010;3(4):388–97.
Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation. 1989;79(4):733–43.
Burke AP, Farb A, Malcom GT, Liang Y, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336(18):1276.
Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, et al. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103(7):934–40.
Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: the pathologists' view. Eur Heart J. 2013;34(10):719–28.
Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30(7):1282–92.
Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47(8 Suppl):C13–8.
Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20(5):1262–75.
Boden WE, O'Rourke RA, Teo KK, Hartigan PM, Maron DJ, Kostuk WJ, et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med. 2007;356(15):1503–16.
Kato K, Yonetsu T, Kim SJ, Xing L, Lee H, McNulty I, et al. Nonculprit plaques in patients with acute coronary syndromes have more vulnerable features compared with those with non-acute coronary syndromes: a 3-vessel optical coherence tomography study. Circ Cardiovasc Imaging. 2012;5(4):433–40.
Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, et al. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116(24):2841–50.
Otsuka F, Finn AV, Virmani R. Do vulnerable and ruptured plaques hide in heavily calcified arteries? Atherosclerosis. 2013;229(1):34–7.
New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108(11):1381–91.
Tintut Y, Patel J, Parhami F, Demer LL. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation. 2000;102(21):2636–42.
Radcliff K, Tang TB, Lim J, Zhang Z, Abedin M, Demer LL, et al. Insulin-like growth factor-I regulates proliferation and osteoblastic differentiation of calcifying vascular cells via extracellular signal-regulated protein kinase and phosphatidylinositol 3-kinase pathways. Circ Res. 2005;96(4):398–400.
Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation. 2008;117(22):2938–48.
Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87(11):1055–62.
Shanahan CM. Inflammation ushers in calcification: a cycle of damage and protection? Circulation. 2007;116(24):2782–5.
Bobryshev YV, Killingsworth MC, Huynh TG, Lord RS, Grabs AJ, Valenzuela SM. Are calcifying matrix vesicles in atherosclerotic lesions of cellular origin? Basic Res Cardiol. 2007;102(2):133–43.
Golub EE. Biomineralization and matrix vesicles in biology and pathology. Semin Immunopathol. 2011;33(5):409–17.
Lardinois D, Weder W, Hany TF, Kamel EM, Korom S, Seifert B, et al. Staging of non-small-cell lung cancer with integrated positron-emission tomography and computed tomography. N Engl J Med. 2003;348(25):2500–7.
Blau M, Nagler W, Bender MA. Fluorine-18: a new isotope for bone scanning. J Nucl Med. 1962;3:332–4.
Blau M, Ganatra R, Bender MA. 18F-fluoride for bone imaging. Semin Nucl Med. 1972;2(1):31–7.
Hawkins RA, Choi Y, Huang SC, Hoh CK, Dahlbom M, Schiepers C, et al. Evaluation of the skeletal kinetics of fluorine-18-fluoride ion with PET. J Nucl Med. 1992;33(5):633–42.
Blake GM, Park-Holohan SJ, Cook GJ, Fogelman I. Quantitative studies of bone with the use of 18F-fluoride and 99mTc-methylene diphosphonate. Semin Nucl Med. 2001;31(1):28–49.
Wootton R, Dore C. The single-passage extraction of 18F in rabbit bone. Clin Phys Physiol Meas. 1986;7(4):333–43.
Hoh CK, Hawkins RA, Dahlbom M, Glaspy JA, Seeger LL, Choi Y, et al. Whole body skeletal imaging with [18F]fluoride ion and PET. J Comput Assist Tomogr. 1993;17(1):34–41.
Cook GJ, Blake GM, Marsden PK, Cronin B, Fogelman I. Quantification of skeletal kinetic indices in Paget's disease using dynamic 18F-fluoride positron emission tomography. J Bone Miner Res. 2002;17(5):854–9.
Installe J, Nzeusseu A, Bol A, Depresseux G, Devogelaer JP, Lonneux M. (18)F-fluoride PET for monitoring therapeutic response in Paget's disease of bone. J Nucl Med. 2005;46(10):1650–8.
Frost ML, Fogelman I, Blake GM, Marsden PK, Cook Jr G. Dissociation between global markers of bone formation and direct measurement of spinal bone formation in osteoporosis. J Bone Miner Res. 2004;19(11):1797–804.
Messa C, Goodman WG, Hoh CK, Choi Y, Nissenson AR, Salusky IB, et al. Bone metabolic activity measured with positron emission tomography and [18F]fluoride ion in renal osteodystrophy: correlation with bone histomorphometry. J Clin Endocrinol Metab. 1993;77(4):949–55.
Hsu WK, Feeley BT, Krenek L, Stout DB, Chatziioannou AF, Lieberman JR. The use of 18F-fluoride and 18F-FDG PET scans to assess fracture healing in a rat femur model. Eur J Nucl Med Mol Imaging. 2007;34(8):1291–301.
Schiepers C, Broos P, Miserez M, Bormans G, De Roo M. Measurement of skeletal flow with positron emission tomography and 18F-fluoride in femoral head osteonecrosis. Arch Orthop Trauma Surg. 1998;118(3):131–5.
Petren-Mallmin M, Andreasson I, Ljunggren O, Ahlstrom H, Bergh J, Antoni G, et al. Skeletal metastases from breast cancer: uptake of 18F-fluoride measured with positron emission tomography in correlation with CT. Skelet Radiol. 1998;27(2):72–6.
Schirrmeister H, Guhlmann A, Kotzerke J, Santjohanser C, Kuhn T, Kreienberg R, et al. Early detection and accurate description of extent of metastatic bone disease in breast cancer with fluoride ion and positron emission tomography. J Clin Oncol. 1999;17(8):2381–9.
Hetzel M, Arslandemir C, Konig HH, Buck AK, Nussle K, Glatting G, et al. F-18 NaF PET for detection of bone metastases in lung cancer: accuracy, cost-effectiveness, and impact on patient management. J Bone Miner Res. 2003;18(12):2206–14.
Even-Sapir E, Metser U, Mishani E, Lievshitz G, Lerman H, Leibovitch I. The detection of bone metastases in patients with high-risk prostate cancer: 99mTc-MDP Planar bone scintigraphy, single- and multi-field-of-view SPECT, 18F-fluoride PET, and 18F-fluoride PET/CT. J Nucl Med. 2006;47(2):287–97.
Beheshti M, Vali R, Waldenberger P, Fitz F, Nader M, Loidl W, et al. Detection of bone metastases in patients with prostate cancer by 18F fluorocholine and 18F fluoride PET-CT: a comparative study. Eur J Nucl Med Mol Imaging. 2008;35(10):1766–74.
Rey C, Combes C, Drouet C, Glimcher MJ. Bone mineral: update on chemical composition and structure. Osteoporos Int. 2009;20(6):1013–21.
Agnese Irkle JLB, Skepper JN, Dweck MR, Joshi FR, Vesey AT, Bennett M, et al. 18F-NaF - a Specific Marker for Vascular Calcification in Atherosclerosis. Circulation. 2013;128:A17385.
Derlin T, Richter U, Bannas P, Begemann P, Buchert R, Mester J, et al. Feasibility of 18F-sodium fluoride PET/CT for imaging of atherosclerotic plaque. J Nucl Med. 2010;51(6):862–5.
Derlin T, Toth Z, Papp L, Wisotzki C, Apostolova I, Habermann CR, et al. Correlation of inflammation assessed by 18F-FDG PET, active mineral deposition assessed by 18F-fluoride PET, and vascular calcification in atherosclerotic plaque: a dual-tracer PET/CT study. J Nucl Med. 2011;52(7):1020–7.
Janssen T, Bannas P, Herrmann J, Veldhoen S, Busch JD, Treszl A, et al. Association of linear (18)F-sodium fluoride accumulation in femoral arteries as a measure of diffuse calcification with cardiovascular risk factors: A PET/CT study. J Nucl Cardiol. 2013;20(4):569–77.
Beheshti M, Saboury B, Mehta NN, Torigian DA, Werner T, Mohler E, et al. Detection and global quantification of cardiovascular molecular calcification by fluoro18-fluoride positron emission tomography/computed tomography–a novel concept. Hell J Nucl Med. 2011;14(2):114–20.
Dweck MR, Jones C, Joshi N, Fletcher AM, Richardson H, White A, et al. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation. 2011. doi:10.1161/CIRCULATIONAHA.111.051052. This important study looks at 18F-fluoride uptake in aortic valve disease.
Dweck MR, Khaw HJ, Sng GK, Luo EL, Baird A, Williams MC, et al. Aortic stenosis, atherosclerosis, and skeletal bone: is there a common link with calcification and inflammation? Eur Heart J. 2013;34(21):1567–74.
Dweck MR, Jenkins WS, Vesey AT, Pringle MA, Chin CW, Malley TS, et al. 18F-sodium fluoride uptake is a marker of active calcification and disease progression in patients with aortic stenosis. Circ Cardiovasc Imaging. 2014;7(2):371–8.
Dweck MR, Chow MW, Joshi NV, Williams MC, Jones C, Fletcher AM, et al. Coronary arterial 18F-sodium fluoride uptake: a novel marker of plaque biology. J Am Coll Cardiol. 2012;59(17):1539–48. We first described the coronary uptake of 18F-fluoride in patients with or without aortic stenosis in this important paper.
Joshi NV, Vesey AT, Williams MC, Shah AS, Calvert PA, Craighead FH, et al. F-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: a prospective clinical trial. Lancet. 2013. doi:10.1016/S0140-6736(13)61754-7. In this paper, we show that 18F-fluoride can identify ruptured and high risk plaques in patients with myocardial infarction and stable angina. Furthermore, we characterise these plaques with intravascular imaging in patients with stable angina and with histology in patients undergoing carotid endartrectomy.
Compliance with Ethics Guidelines
Conflict of Interest
Nikhil V, Joshi, Alex Vesey, David E, Newby, and Marc R. Dweck declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Nuclear Cardiology
Rights and permissions
About this article

Cite this article
Joshi, N.V., Vesey, A., Newby, D.E. et al. Will 18F-Sodium Fluoride PET-CT Imaging Be the Magic Bullet for Identifying Vulnerable Coronary Atherosclerotic Plaques?. Curr Cardiol Rep 16, 521 (2014). https://doi.org/10.1007/s11886-014-0521-4
Published:
DOI: https://doi.org/10.1007/s11886-014-0521-4