
Abstract
Takayasu arteritis (TA) is 1 of the 2 main causes of large vessel vasculitides (LVV), giant cell arteritis being the other. LVV can also develop in various other systemic diseases. In TA, a wide variety of symptoms result from vascular stenoses, occlusions, and dilation. Aneurysms may develop and may occasionally dissect or rupture. Disease activity can sometimes be difficult to assess clinically. Diagnostic modalities also have their shortcomings. Often, acute phase reactants do not accurately detect disease activity. Available vascular imaging modalities may be acceptable in defining vascular anatomy, but are notoriously inaccurate in delineating vascular inflammation. Glucocorticoids remain the cornerstone of therapy in TA, in spite of foreseeable long term side effects. In addition, several steroid-sparing agents are also being used, often based on promising results from small uncontrolled studies. Rarely, endovascular revascularization procedures are necessary. Resection of critical-sized aortic aneurysms and repair of aortic dissections are occasionally warranted as lifesaving procedures. The long term outcome of surgical intervention is often unfavorable and relapses are not uncommon. In addition to TA, other less commonly encountered causes of LVV are also briefly discussed in this review.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Takayasu arteritis (TA) is 1 of the 2 main causes of large vessel vasculitides (LVV), giant cell arteritis (GCA) being the other. TA is typically seen in younger patients with age of onset less than 40 years, whereas GCA normally starts after the age of 50 years. According to some experts, TA and GCA may fall within the spectrum of the same disease [1•]. Both TA and its therapy can lead to significant morbidity and premature mortality. Clinical manifestations of TA are quite variable, ranging from tissue ischemia due to vascular stenosis and occlusion, and to aneurysm formation that may occasionally rupture or dissect. Systemic symptoms (fever, malaise, weight loss, night sweats, polyarthralgia or arthritis) may predominate at the onset of TA. Hence, it is not uncommon for the unsuspecting clinician to miss the underlying diagnosis, if proper vigilance is not exercised in obtaining a comprehensive history and performing a meticulous physical examination, particularly that of the vascular system—assessment of pulse and blood pressure in all 4 extremities, and listening for bruits and for an aortic regurgitation murmur. However, despite all diligence, clinical evaluation can be notoriously unreliable in determining disease activity, and silent disease progression is not uncommon. Furthermore, progressive vascular stenosis or occlusion does not automatically signify ongoing vasculitis, but could simply indicate advancing fibrosis. Likewise, aneurysm enlargement could also be a result of hemodynamic compromise, exacerbated by poorly controlled hypertension.
Diagnostic modalities of TA have their deficiencies. At the onset, the acute phase reactants can be normal in spite of active disease, and they are also unreliable biomarkers of remission or relapse. Activity (vascular inflammation, which is potentially reversible with immunosuppressive medications) is often difficult to differentiate from damage (which is cumulative and potentially irreversible). Available vascular imaging modalities may be acceptable in defining vascular anatomy (luminal narrowing, occlusion, ectasia, or dissection) [2], and in demonstrating wall thickening and calcification, but are notoriously inaccurate in delineating vascular inflammation that could potentially guide immunosuppressive therapy.
Glucocorticoids have remained the backbone of therapy of TA, in spite of anticipated long term side effects. In addition, several steroid-sparing agents (conventional disease modifying agents and biologic response modifiers) are also being used, often based on encouraging results from small uncontrolled studies. Rarely, endovascular revascularization procedures are necessary. Resection of critical-sized aortic aneurysms and repair of aortic dissections are occasionally warranted as lifesaving procedures. The long term outcome of surgical intervention is often unfavorable and relapses are not rare, especially when TA is active perioperatively. Close vigilance during and post-treatment is needed to monitor for relapses.
In addition to TA, various other less commonly encountered causes of LVV (Table 1) are discussed briefly in this article, as detailed description is beyond the scope of this review.
Takayasu Arteritis
Takayasu arteritis (TA) is a chronic idiopathic granulomatous LVV affecting the aorta and its first order branches [3]. It tends to affect patients up to 50 years of age with a striking female predominance (80 %–90 %). The age of onset is usually between 10 and 40 years [4, 5] and the racial profile is diverse. In the US, Caucasian patients outnumber Asian patients, though worldwide, the greatest prevalence is in Asians [6–8]. In Japan, the annual incidence is about 150 new cases/year [9] whereas in the US and Europe, the annual incidence is 1–3 new cases/million population [5]. HLA-Bw52 and HLA-B39.2 association have been described in some studies, suggesting an immunogenetic predisposition [10].
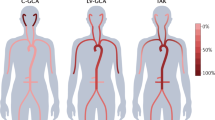
In TA, inflammation may be localized to a portion of the thoracic or abdominal aorta and its branches, or may involve the entire vessel. Although there is considerable variability in disease expression [11], the initial vascular lesion often starts in the left subclavian artery and subsequently spreads to involve the left common carotid, left vertebral, brachiocephalic, right subclavian, right vertebral, and right common carotid arteries. Thoracic aorta is commonly affected, whereas abdominal aorta and pulmonary arteries [4, 12] are involved in approximately 50 % of patients. Abdominal aorta and/or renal arteries are more commonly affected in Indian patients [3].
Reactive hyperplasia and fibrosis causes thickening of the arterial wall, whereas the proximal aorta may sometimes become dilated due to inflammatory injury. Narrowing, occlusion, or dilation of involved portions of the arteries in varying degrees results in a wide variety of manifestations [13].
Clinical Features
Symptoms resulting from systemic inflammation such as fever, weight loss, fatigue, malaise, myalgia and arthralgia are common at the onset of disease. Arterial inflammation may also cause headache, thoracic pain, and carotidynia. In addition, features related to arterial compromise (limb claudication, abdominal and cardiac angina, heart failure, dizziness, symptoms from an enlarging aneurysm, dissection, transient ischemic attack, or stroke) can occur. However, it is critical to note that asymptomatic progression is not at all uncommon.
On physical examination, peripheral pulses may be feeble or absent. Patients may develop new onset hypertension, or asymmetrical blood pressure recordings in the extremities (between both arms or between the upper and the lower extremities). It is important to listen for vascular bruits and palpate for superficial arterial tenderness (eg, carotids). Careful cardiac auscultation may identify a previously undetected aortic regurgitation murmur. It is also advisable to perform ophthalmoscopy and a urinalysis to identify end organ damage attributable to hypertension.
Though clinical examination has excellent specificity, it has been found to have poor sensitivity [14]. In a study of 100 patients (2/3 with TA and 1/3 with GCA) undergoing standardized physical examination and angiography, individual physical examination findings had poor sensitivity (14 %–50 %) but high specificity (71 %–98 %) to determine the arteriographic lesions [14]. Considering all physical examination findings combined, at least 30 % of angiographic lesions were missed [14]. So abnormal physical examination findings are highly associated with the presence of arterial lesions, but normal findings do not exclude the possibility of arterial disease. This supports the rationale for serial angiographic assessment to monitor arterial disease in patients with established TA.
Pathology
TA affects large and medium-sized elastic arteries resulting in circumferential thickening of the vessel (Fig. 1c and f). In the acute phase, there is florid inflammation that predominantly affects both the media and the adventitia. Inflammatory cells are composed of a mixture of lymphocytes, plasma cells and macrophages. Giant cells are common (Fig. 1c). The intima shows reactive hyperplasia that, when florid, can result in ostial stenosis of the branch vessels arising from the aorta. This reactive intima is also predisposed to secondary atherosclerosis. In the chronic lesions, the media shows scarring with fragmentation and loss of elastic lamellae often accompanied by marked fibrosis of the adventitia. The resulting severe adventitial thickening is thought to prevent aneurysmal dilation of the aorta in TA.

Histopathology of large vessel vasculitis: the panel shows (a) photomicrographs of a normal aorta, (b) giant cell arteritis, and (c) Takayasu arteritis, with corresponding sections stained with Movat pentachrome (d, e, and f). The images are all taken at the same low magnification: (a) The intima and adventitial layers of a normal aorta are very thin; (d) The media shows multiple orderly layers of elastic lamellae stained black with Movat; (B) A case of giant cell aortitis shows medial necrosis surrounded by mononuclear inflammatory cells and multinucleated giant cells (inset); (e) The elastic lamellae are disrupted in areas of inflammation but remain visible in the area of medial necrosis; (c) A case of Takayasu arteritis shows inflammation in the media and adventitia with predominantly mononuclear cells and occasional multinucleated giant cells (inset); (f) The media shows disruption and collapse of the elastic layer. Note that the intima in both aortitis cases is thicker than the media due to reactive hyperplasia and fibrosis. (e and f) The intimal hyperplasia appears blue green on Movat due to increased ground substance. The adventitia shows fibrous thickening which becomes progressively more prominent in Takayasu arteritis. (e and f) The dense adventitial fibrosis is delineated by the yellow staining of collagen on Movat
Diagnosis
In addition to a careful history and physical examination as described above, diagnosis of TA should be based on laboratory studies (acute phase reactants), and imaging studies. Rarely, histology of the resected vessel is also available, and may help establishing the diagnosis and assess its severity.
Laboratory Studies: Acute Phase Reactants (ESR and CRP)
Acute phase reactants (ESR and CRP) are commonly ordered, along with complete blood count and comprehensive metabolic panel, not only to assess disease activity but also to evaluate complications of therapy or evidence of end organ damage. Sometimes periodic laboratory testing is needed to assess relevant co-morbidities, eg, fasting lipids, blood glucose, etc.
About 25 % of patients have a normal acute phase response even in the presence of active disease [15, 16]. It has been found that about 46 % patients have normal ESR and CRP along with new MRA changes, whereas others have elevated acute phase reactants in the setting of an unchanged MRI. So even if acute phase responses are normal, we must maintain ongoing vigilance for evidence of active disease. On the other hand, if they are elevated, we must still rule out other causes (eg, infection), to avoid escalation of immunosuppression purely based on these laboratory abnormalities.
In addition to ESR and CRP, a whole array of biomarkers have been studied: interleukin(IL)-6, serum amyloid A, fibrinogen, complement split fragments, B-cell activating factor (BAFF), interleukin(IL)-12, matrix metalloproteinase 9 (MMP-9), and pentraxin 3 (PTX3). To date, there has been no established biomarker for accurate assessment of disease activity in TA. This task is even more arduous as the definition of a gold standard for assessing disease activity in TA is unclear.
Imaging Modalities [17, 18]
As in GCA, the available imaging modalities are computerized tomographic angiography (CTA), magnetic resonance angiography (MRA) (Figs. 2a, b, and 3), positron emission tomography with or without CT scan (PET/PET-CT), Doppler ultrasound and catheter directed angiography (CDA) (Fig. 2c-f). Considerable controversies exist about the modality that is best for diagnosis and for follow-up, one that is most influenced by therapy and one that best predicts complications. Also, there is no consensus as to how often these imaging studies should be repeated. The pros and cons of the different imaging modalities for large vessel vasculitides have been discussed in detail in the previous article: on GCA (here it says 6–8). In addition, similar to LV-GCA, the same potential pitfalls apply (here it says 7) for vascular imaging in TA. Also, there are uncertainties about the significance of the changes that occur in wall thickening and enhancement after initiation of treatment. Therefore, only the development of a new lesion in a previously unaffected vascular territory is regarded as clear evidence of active disease and this should prompt a thorough evaluation of new but asymptomatic lesions in other territories. This can be achieved by periodic imaging of the aorta and all its first order branches, from the neck to the pelvis. Other dedicated imaging studies (eg, cerebral and/or coronary circulation) may be required based on the presentation or history of past involvement in those areas.

Large vessel vasculitis: (a) MRA aortic arch: 3D-reconstruction of a contrast-enhanced magnetic resonance angiogram of the thoracic aorta and proximal arch branch vessels demonstrates a widely patent innominate artery, severe stenosis of the left common carotid artery origin (white arrow), and total occlusion of the left subclavian artery origin (white arrowhead). The left subclavian artery fills via retrograde flow from the left vertebral artery; (b) MRA aortic arch (same patient, 2 years later): 3D-reconstruction of a contrast-enhanced magnetic resonance angiogram of the thoracic aorta and proximal arch branch vessels again demonstrates a widely patent innominate artery, and total occlusion of the left subclavian artery origin (white arrowhead). However, there has also been interval occlusion of the left common carotid artery (the expected origin is denoted by white arrow, and the dashed line indicates the expected course of the left common carotid artery); (c) Digital subtraction angiography (DSA) of the aortic arch demonstrating a widely patent innominate artery (white arrow); (d) DSA of the aortic arch, moments later, demonstrating faint retrograde filling of the left vertebral artery (between arrowheads), and left subclavian artery (arrows); (e) DSA of the aortic arch (without contrast) demonstrating successful positioning of a stent in the proximal left subclavian artery (arrow); (f) DSA of the aortic arch demonstrating widely patent left subclavian and left vertebral arteries following stent placement

Takayasu arteritis: (a) 3D-reconstruction of a contrast-enhanced MRA demonstrating absence of the left renal artery (prior occlusion), and an apparent severe stenosis of the proximal right renal artery (white arrow); (b) 3D-phase-contrast, axial image at the level of the renal arteries. There is no left renal artery (white asterisk) as a result of prior total occlusion. The right renal artery is patent, though lack of bright signal in the proximal portion (between white arrows) confirms hemodynamically severe renal artery stenosis; (c) Coronal source image from a contrast-enhanced MRA demonstrating the normal sized and normally enhancing right kidney. An atrophied left kidney is present and has minimal cortical enhancement from tiny collateral vessels (not seen)
There are no published comparative studies to guide us about the optimal imaging modality. The challenge arises as tissue correlates of the imaging findings are rarely available. Cost and accessibility must be weighed against the concerns of radiation and contrast toxicity and other patient factors. Vascular imaging is recommended if a relapse is suspected, or, if asymptomatic, at least annually, to exclude disease progression, or worsening due to mechanical factors.
Disease Activity Assessment
Currently, a reliable tool to assess disease activity is an unmet need in TA. Traditionally, the NIH (Kerr) criteria have been used, which defines activity as new development or worsening of at least 2 of the following features: (a) constitutional symptoms and signs, (b) elevated acute phase reactants, (c) symptoms and signs of vascular insufficiency, and (d) new vascular lesions on serial imaging studies [15]. Newer validated instruments of disease activity assessment are being described, such as DEI-TAK criteria [19] and Indian Takayasu Activity Score (ITAS) [20]. There are ongoing efforts via OMERACT to try to refine disease activity measures for TA.
Unlike GCA (where temporal artery biopsies are routinely performed), vascular tissue is rarely available in TA, and can only be obtained when vascular surgery is being performed. The flaws of the available disease activity measures become clear if we bear in mind that surgery is generally undertaken during periods when the inflammatory disease is felt to be in remission, and the vascular bypass is likely to connect into a ‘normal’ appearing vessel segment. Despite these factors, approximately, half of the surgical specimens show active vasculitis [15, 21]. A recent retrospective study showed that only 20 % of patients with active aortitis on histopathology had disease that was thought to be clinically active at the time of surgery [22]. So, even carefully considering all the available elements of assessment, undoubtedly there are major limitations in evaluating disease activity in TA.
Pregnancy and TA
The main complications of TA that can raise concerns during pregnancy are aortic regurgitation, cardiac failure, renal artery stenosis, and poorly controlled hypertension. In the absence of these complications, the risk of well controlled disease on pregnancy is not substantial. Appropriate contraceptive measures should be adopted and pregnancy should always be planned. Patients can be provided some reassurance that with close maternal and fetal monitoring, successful pregnancy outcomes have been reported and fetal outcomes are not necessarily unfavorable [16, 23].
Management
Medical Treatment
Corticosteroids have been the mainstay of therapy for active TA [16]. The classic approach has been prednisone 40–60 mg/day (1 mg/Kg/day) for about a month, and then reducing the dose by 5 mg/week until 20 mg/day is reached, and then by 2.5 mg/week until 10 mg/day is reached. Then the dose is usually tapered by 1 mg/month until discontinuation [16]. Glucocorticoids induce remission in about 60 % of patients, but relapses do occur in the majority (>50 %) during steroid taper [15]. More recent studies also reinforce the fact that disease control with prednisone alone is infrequent (≤20 % patients), and that majority (66 %–84 %) of patients do require other immunosuppressive agents [16, 24, 25•]. In addition, adverse effects of glucocorticoids are common, including adrenal suppression, opportunistic infections, osteoporosis, hypertension, diabetes, accelerated atherosclerosis, steroid myopathy, cataracts, glaucoma, sleep disturbance, mood disturbances, and even psychosis. So, patients initiating treatment of TA often need other medications to counteract the side effects of glucocorticoids.
In an attempt to reduce the risk of expected side effects of long term glucocorticoids, various other immunosuppressive agents have been tried. In small uncontrolled studies and in case series, all of the following agents have been reported to be of some benefit, but none of these agents have been evaluated in any randomized control trial: methotrexate [26], azathioprine [27], mycophenolate mofetil [28, 29], leflunomide [30], and cyclophosphamide [16]. In relapsing disease [16], experts recommend increasing prednisone dose by 10 mg/day over the previous effective dose, and then add a second oral agent, either methotrexate or azathioprine. Mycophenolate mofetil is considered third line, whereas cyclophosphamide is generally reserved for immediately life-threatening disease, as it should preferably be avoided in this young, typically female population. Often, at the initial presentation, a second immunosuppressive agent is concomitantly started along with a glucocorticoid, given the high likelihood of relapse that had been identified in most series. However, it is unclear whether this should be done in every patient or only if there is evidence of arterial damage at baseline. Though there are no clear guidelines, this approach would seem appropriate in selected patients.
One study indicated that low dose aspirin use was associated with a lower frequency of ischemic events [31]. In the absence of any controlled trial data, it would still appear reasonable to have a low threshold for prescribing aspirin for all patients, and probably it should be considered routinely for those with arterial stenosis or occlusion.
Hypertension is a particularly important complication in patients with TA. New onset hypertension can indicate development of renal artery stenosis, which can represent active disease (in up to 80 % of patients). Hypertension can also develop from aortic stiffness due to long standing disease. It may present with headaches or with evidence of end-organ damage, eg, congestive heart failure, hypertensive retinopathy, stroke, and renal disease. It may go unrecognized, as patients can have central aortic hypertension on catheter directed angiography, which is missed on conventional blood pressure measurement due to peripheral arterial (subclavian and brachial) stenosis. Pressure gradient to the arm can be measured and be used as a surrogate to monitor a patient’s central aortic blood pressure. Anti-hypertensive therapy must be judiciously introduced to prevent worsening of ischemic symptoms. Renal artery revascularization is considered in refractory patients. Ongoing monitoring of blood pressure remains a challenging dilemma, given the absence of a reliable method of noninvasive assessment of central aortic pressure. On a regular basis, evidence of end organ damage should be monitored for: urinalysis, retinal examination, echocardiogram, and at least in selected patients, periodic catheter directed angiography.
Emerging Treatment Options
Trials involving tumor necrosis factor-α (TNF) antagonists have been conducted in TA [32, 33]. Twenty-five steroid and methotrexate/azathioprine-refractory TA patients were treated with infliximab or etanercept [33]. The median infliximab dose was 5 mg/Kg every 6 weeks. Fifteen of these patients were able to achieve remission off prednisone (60 %) and 7 went into remission on low dose prednisone (<10 mg/day) (28 %). The median prednisone dose prior to introduction of anti-TNF therapy was 19 mg/day (5–50 mg), which could be discontinued after initiating anti-TNF therapy. None of the patients on azathioprine or methotrexate at the time of institution of anti-TNF therapy were able to taper or discontinue that therapy. Eight out of 9 patients that discontinued infliximab for various reasons had a relapse within a median duration of 5 months. Four relapses occurred on anti-TNF treatment, 3 of whom were controlled with increased dose of anti-TNF therapy. Similar results with anti-TNF agents have been shown in 3 other studies [34•, 35, 36]. However, despite these encouraging reports, a randomized controlled trial is still required to establish efficacy and safety of anti-TNF therapy in TA.
In recent years, some case reports and case series indicate the efficacy of tocilizumab in TA [37•, 38•]. Again, randomized controlled trials would be necessary to demonstrate its efficacy. Currently, a randomized controlled trial using abatacept is ongoing. Also, there have been a few case reports indicating the effectiveness of rituximab [39••, 40, 41••, 42, 43].
Hence, though the signals from small preliminary studies are encouraging, in the absence of data from any randomized controlled trial, there is no unequivocally proven biologic therapy for TA at this time.
Vasculitis Mimics
Before labeling a patient to have “refractory” TA, and hence, escalating immunosuppressive therapy, it is important to take a step back and reconsider the diagnosis. It would be important to make sure that a “vasculitis mimic” has not been missed. Differential diagnoses should include infections that are known to involve blood vessels, eg, tuberculosis, syphilis, HIV infection, or bacterial infections of the arterial wall leading to mycotic aneurysms. Noninflammatory conditions would include: (a) atherosclerosis (b) thromboembolic diseases, (c) genetic disorders (Marfan syndrome, Ehlers-Danlos syndrome type IV, Loeys-Dietz syndrome, Grange syndrome), (d) congenital disorders (coarctation of the aorta, Turner syndrome, Williams syndrome), and (e) conditions with unknown etiology (fibromuscular dysplasia, segmental arterial mediolysis). When dealing with infectious etiologies, we need to treat the underlying infection, whereas with other vasculitis mimics the treatment should be geared toward the underlying disease when possible. Therefore, more aggressive immunosuppression should only be reserved for truly “refractory” cases of primary LVV. Even when the diagnosis of TA is definite, sometimes the manifestations could be more due to vascular complications, tissue damage, drug adverse effects, infection, or other concomitant illness, for which intensifying immunosuppressive therapy would not be appropriate, and management should be directed toward addressing the underlying problem.
Nonpharmacologic Measures
Smoking in the context of an inflammatory vascular disease is strongly discouraged. Patients should be repeatedly reminded of this, and referred for appropriate counseling if necessary. Diet is important for bone health and cardiovascular health in general, and also for maintaining a healthy body weight while on glucocorticoids, but it has not been shown to have a clear role in affecting the disease course as such. Exercise is also important in patients with TA, in particular to reduce some of the glucocorticoid side effects such as bone loss, weight gain, etc. Moreover, exercise may potentially improve exercise tolerance in patients with claudication.
Surgical Treatment
The outcomes of revascularization procedures are often unfavorable. Affected vessels do not necessarily need to be ‘unblocked’ as most patients develop these stenoses over a long period of time, and collateral flow is generally well developed. Hence, patients do not usually develop distal ischemia or infarction. Moreover, there is a high failure rate with revascularization procedures in general; so it is important to carefully select the patients who would likely benefit from these interventions.
Vascular lesions frequently involve long segments, and are often fibrotic and even calcified. So they are not generally amenable to endovascular procedures, and careful selection of lesions is essential. Even in the carefully selected cases, endovascular procedures often result in suboptimal outcomes, as after initial restoration of vascular patency, restenoses are quite common. Some studies suggest that over 50 % will restenose over time [44, 45]. It is not clear what the impact of newer approaches are, such as drug eluting stents, covered stents, or stent grafts [46], although there is some evidence in small numbers of patients that they might be somewhat better than the traditional endovascular procedures.
Surgical intervention for aortic disease typically includes aortic root reconstruction and vascular bypass grafting. A bypass graft should ideally take origin from the aortic root and not a branch vessel. For example, if the graft connects a subclavian artery to a carotid artery, and the subclavian artery later becomes occluded (as part of progression of disease), then both vessels are likely to be lost. When a surgical repair is absolutely indicated, it is best performed during a period of remission if possible. A French study identified that the overall 5-year complication rate was about 44 % [47]. Approximately a third of patients who had surgical procedures needed secondary intervention as opposed to endovascular procedures, where 50 % needed secondary intervention. The risk of failure was about 7-fold higher if there was evidence of inflammation at the time of the procedure [47].
Prognosis
TA is considered to be a rare but serious disease with a chronic relapsing-remitting course. The disease, as well as the adverse effects of long term glucocorticoids and other immunosuppressive agents, often causes significant morbidity and disability [16, 48]. These young, predominantly female patients can also die prematurely from complications of this disease. In a cohort study of 126 TA patients, mortality was increased compared with the general population (standardized mortality ratio: 3.0) [25•]. Limited data on the potential disease-modifying effects of TNF inhibitors [16, 33, 34•] and other biologic agents are encouraging and merit further evaluation.
Other Causes of LVV
Large vessel involvement occurs in approximately 30 % of patients with Behçet’s disease [49]. Perivascular and endovascular inflammation causes hemorrhage, stenosis, aneurysm formation, and both arterial and venous thrombosis. The disease follows a relapsing-remitting course and is usually treated with immunosuppressive agents, though surgery is sometimes necessary [50]. Carotid, pulmonary, aortic, iliac, femoral, and popliteal arteries are often affected, whereas cerebral and renal arteries, much less so. Aneurysms involving the large proximal branches of the pulmonary artery are the commonest and the most characteristic of the vascular lesions in Behçet’s disease. These lesions are distinctly uncommon in other forms of LVV. Patients often present with hemoptysis. Cough, dyspnea, fever, and pleuritic pain can also occur [51, 52]. Anticoagulation due to a misdiagnosis of pulmonary embolism can lead to devastating consequences [51]. Ventilation-perfusion scans are often not helpful, but pulmonary angiography confirms the diagnosis.
Aortitis has also been described in rheumatoid arthritis [53], ankylosing spondylitis [54, 55], Cogan’s syndrome (10 % cases) [56], and relapsing polychondritis [57, 58] (Table 1). A newly recognized disorder, IgG4-related systemic disease, has also been known to cause aortitis, peri-aortitis, and retroperitoneal fibrosis [59•]. Detailed description of these conditions is beyond the scope of this review.
Conclusions
TA is a chronic large vessel vasculitis, mostly seen in young females, that predominantly affects the aorta and its major branches. TA and the required immunosuppressive therapy cause considerable morbidity and mortality. Consequences of vascular stenoses, occlusions, and less commonly vascular dilation account for the typical clinical presentation; the latter can sometimes lead to aneurysmal rupture or dissection. Though the importance of a comprehensive history and a thorough physical examination cannot be over-emphasized, clinical assessment is frequently inaccurate in evaluating disease activity, which may sometimes progress silently. Also, the diagnostic modalities currently used are unsatisfactory. Acute phase reactants can be normal when disease is active and elevated due to other causes when disease is quiet. Available imaging modalities are also inadequate, and though they are reasonably precise in defining vascular anatomy, they often do not help assessing disease activity accurately or guide treatment recommendations. Progressive vascular narrowing could simply indicate perpetuation of fibrosis, and aneurysm enlargement could solely result from altered hemodynamics, resulting from uncontrolled hypertension. Glucocorticoids are the therapeutic agents of choice for most patients, though adverse effects are almost inevitable. Hence, in spite of absence of large randomized studies, several steroid-sparing agents are currently being tried, based on their proven effectiveness in several small uncontrolled trials. Surgical treatment, which should preferably be avoided, is also needed at times based on specific circumstances. Results of surgery are often unsatisfactory if TA is active perioperatively. When TA is felt to be in remission, the short term results of revascularization procedures may be acceptable, but relapses occur quite often when patients are followed over several years. It is imperative that better biomarkers and better imaging modalities be devised for accurate evaluation of ongoing vascular inflammation, so that the effect of therapy can be precisely quantified. A more detailed understanding of the pathogenesis of TA is likely to provide new targets for therapy.
LVV has also been associated with Behçet’s disease, rheumatoid arthritis, ankylosing spondylitis, Cogan’s syndrome, relapsing polychondritis, and IgG4-related systemic disease. Detailed description of these conditions is beyond the scope of this review.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Takayasu arteritis and giant cell arteritis: a spectrum within the same disease? Medicine. 2009;88:221–6. This retrospective review led to the introduction of the hypothesis that TA and GCA may represent different phenotypes within the spectrum of a single disorder.
Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum. 2002;46:1634–42.
Hata A, Noda M, Moriwaki R, Numano F. Angiographic findings of Takayasu arteritis: new classification. Int J Cardiol. 1996;54(Suppl):S155–63.
Lupi-Herrera E, Sanchez-Torres G, Marcushamer J, Mispireta J, Horwitz S, Vela JE. Takayasu's arteritis. Clinical study of 107 cases. Am Heart J. 1977;93:94–103.
Arend WP, Michel BA, Bloch DA, Hunder GG, Calabrese LH, Edworthy SM, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33:1129–34.
Dabague J, Reyes PA. Takayasu arteritis in Mexico: a 38-year clinical perspective through literature review. Int J Cardiol. 1996;54(Suppl):S103–9.
Hall S, Barr W, Lie JT, Stanson AW, Kazmier FJ, Hunder GG. Takayasu arteritis. A study of 32 North American patients. Medicine. 1985;64:89–99.
Ishikawa K. Natural history and classification of occlusive thromboaortopathy (Takayasu’s disease). Circulation. 1978;57:27–35.
Koide K. Takayasu arteritis in Japan. Heart Vessels. 1992;7(Suppl):48–54.
Kimura A, Kitamura H, Date Y, Numano F. Comprehensive analysis of HLA genes in Takayasu arteritis in Japan. Int J Cardiol. 1996;54(Suppl):S61–9.
Cid MC, Font C, Coll-Vinent B, Grau JM. Large vessel vasculitides. Curr Opin Rheumatol. 1998;10:18–28.
Lupi E, Sanchez G, Horwitz S, Gutierrez E. Pulmonary artery involvement in Takayasu’s arteritis. Chest. 1975;67:69–74.
Sharma BK, Jain S, Sagar S. Systemic manifestations of Takayasu arteritis: the expanding spectrum. Int J Cardiol. 1996;54(Suppl):S149–54.
Grayson PC, Tomasson G, Cuthbertson D, Carette S, Hoffman GS, Khalidi NA, et al. Association of vascular physical examination findings and arteriographic lesions in large vessel vasculitis. J Rheumatol. 2012;39:303–9.
Kerr GS, Hallahan CW, Giordano J, Leavitt RY, Fauci AS, Rottem M, et al. Takayasu arteritis. Ann Intern Med. 1994;120:919–29.
Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis Rheum. 2007;56:1000–9.
Kermani TA, Warrington KJ. Recent advances in diagnostic strategies for giant cell arteritis. Curr Neurol Neurosci Rep. 2012;12:138–44.
Blockmans D, Bley T, Schmidt W. Imaging for large-vessel vasculitis. Curr Opin Rheumatol. 2009;21:19–28.
Aydin SZ, Yilmaz N, Akar S, Aksu K, Kamali S, Yucel E, et al. Assessment of disease activity and progression in Takayasu’s arteritis with Disease Extent Index-Takayasu. Rheumatology. 2010;49:1889–93.
Misra R, Danda D, Rajappa SM, Ghosh A, Gupta R, Mahendranath KM, et al. Development and initial validation of the Indian Takayasu Clinical Activity Score (ITAS2010). Rheumatology. 2013;52:1795–801.
Lagneau P, Michel JB, Vuong PN. Surgical treatment of Takayasu’s disease. Ann Surg. 1987;205:157–66.
Clifford A, Clark T, Johnston D, Petterson G, Roselli E, Rodriguez E, et al. Large Vessel vasculitis: estimating disease activity in patients with inflammatory thoracic aortic aneurysms. Arthritis Rheum. 2013;65:S10. Abstract.
Wong VC, Wang RY, Tse TF. Pregnancy and Takayasu’s arteritis. Am J Med. 1983;75:597–601.
Bicakcigil M, Aksu K, Kamali S, Ozbalkan Z, Ates A, Karadag O, et al. Takayasu's arteritis in Turkey—clinical and angiographic features of 248 patients. Clin Exp Rheumatol. 2009;27(1 Suppl 52):S59–64.
Schmidt J, Kermani TA, Bacani AK, Crowson CS, Cooper LT, Matteson EL, et al. Diagnostic features, treatment, and outcomes of Takayasu arteritis in a US cohort of 126 patients. Mayo Clin Proc. 2013;88:822–30. Report of a large cohort of patients with TA enrolled in a single center over 25 years.
Hoffman GS, Leavitt RY, Kerr GS, Rottem M, Sneller MC, Fauci AS. Treatment of glucocorticoid-resistant or relapsing Takayasu arteritis with methotrexate. Arthritis Rheum. 1994;37:578–82.
Valsakumar AK, Valappil UC, Jorapur V, Garg N, Nityanand S, Sinha N. Role of immunosuppressive therapy on clinical, immunological, and angiographic outcome in active Takayasu’s arteritis. J Rheumatol. 2003;30:1793–8.
Shinjo SK, Pereira RM, Tizziani VA, Radu AS, Levy-Neto M. Mycophenolate mofetil reduces disease activity and steroid dosage in Takayasu arteritis. Clin Rheumatol. 2007;26:1871–5.
Goel R, Danda D, Mathew J, Edwin N. Mycophenolate mofetil in Takayasu’s arteritis. Clin Rheumatol. 2010;29:329–32.
de Souza AW, da Silva MD, Machado LS, Oliveira AC, Pinheiro FA, Sato EI. Short-term effect of leflunomide in patients with Takayasu arteritis: an observational study. Scand J Rheumatol. 2012;41:227–30.
de Souza AW, Machado NP, Pereira VM, Arraes AE, Reis Neto ET, Mariz HA, et al. Antiplatelet therapy for the prevention of arterial ischemic events in Takayasu arteritis. Circ J. 2010;74:1236–41.
Hoffman GS, Merkel PA, Brasington RD, Lenschow DJ, Liang P. Anti-tumor necrosis factor therapy in patients with difficult to treat Takayasu arteritis. Arthritis Rheum. 2004;50:2296–304.
Molloy ES, Langford CA, Clark TM, Gota CE, Hoffman GS. Anti-tumour necrosis factor therapy in patients with refractory Takayasu arteritis: long-term follow-up. Ann Rheum Dis. 2008;67:1567–9.
Schmidt J, Kermani TA, Bacani AK, Crowson CS, Matteson EL, Warrington KJ. Tumor necrosis factor inhibitors in patients with Takayasu arteritis: experience from a referral center with long-term follow-up. Arthritis Care Res. 2012;64:1079–83. Largest published series of TA patients demonstrating efficacy of treatment with TNF inhibitors.
Mekinian A, Neel A, Sibilia J, Cohen P, Connault J, Lambert M, et al. Efficacy and tolerance of infliximab in refractory Takayasu arteritis: French multi-centre study. Rheumatology. 2012;51:882–6.
Quartuccio L, Schiavon F, Zuliani F, Carraro V, Catarsi E, Tavoni AG, et al. Long-term efficacy and improvement of health-related quality of life in patients with Takayasu’s arteritis treated with infliximab. Clin Exp Rheumatol. 2012;30:922–8.
Unizony S, Arias-Urdaneta L, Miloslavsky E, Arvikar S, Khosroshahi A, Keroack B, et al. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res. 2012;64:1720–9. Largest published case series of tocilizumab therapy in large-vessel vasculitis.
Abisror N, Mekinian A, Lavigne C, Vandenhende MA, Soussan M, Fain O. Tocilizumab in refractory Takayasu arteritis: a case series and updated literature review. Autoimmun Rev. 2013;12(12):1143–9. Report on a compilation of 44 cases reviewing the efficacy of tocilizumab in refractory Takayasu arteritis.
Clifford A, Hoffman GS. Recent advances in the medical management of Takayasu arteritis: an update on use of biologic therapies. Curr Opin Rheumatol. 2014;26:7–15. A nice review on the use of biologic therapies in TA.
Keser G, Direskeneli H, Aksu K. Management of Takayasu arteritis: a systematic review. Rheumatology. 2013.
Unizony S, Stone JH, Stone JR. New treatment strategies in large-vessel vasculitis. Curr Opin Rheumatol. 2013;25:3–9. A very elegant review of biologic response modifiers in the treatment of large-vessel vasculitis.
Ernst D, Greer M, Stoll M, Meyer-Olson D, Schmidt RE, Witte T. Remission achieved in refractory advanced Takayasu arteritis using rituximab. Case Rep Rheumatol. 2012;2012:406963.
Hoyer BF, Mumtaz IM, Loddenkemper K, Bruns A, Sengler C, Hermann KG, et al. Takayasu arteritis is characterised by disturbances of B cell homeostasis and responds to B cell depletion therapy with rituximab. Ann Rheum Dis. 2012;71:75–9.
Kim YW, Kim DI, Park YJ, Yang SS, Lee GY, Kim DK, et al. Surgical bypass vs endovascular treatment for patients with supra-aortic arterial occlusive disease due to Takayasu arteritis. J Vasc Surg. 2012;55:693–700.
Lee G, Jeon P, Do YS, Sung K, Kim DI, Kim YW, et al. Comparison of outcomes between endovascular treatment and bypass surgery in Takayasu arteritis. Scand J Rheumatol. 2013.
Qureshi MA, Martin Z, Greenberg RK. Endovascular management of patients with Takayasu arteritis: stents versus stent grafts. Semin Vasc Surg. 2011;24:44–52.
Saadoun D, Lambert M, Mirault T, Resche-Rigon M, Koskas F, Cluzel P, et al. Retrospective analysis of surgery versus endovascular intervention in Takayasu arteritis: a multicenter experience. Circulation. 2012;125:813–9.
Maksimowicz-McKinnon K, Hoffman GS. Takayasu arteritis: what is the long-term prognosis? Rheum Dis Clin N Am. 2007;33:777–86. vi.
Koc Y, Gullu I, Akpek G, Akpolat T, Kansu E, Kiraz S, et al. Vascular involvement in Behcet’s disease. J Rheumatol. 1992;19:402–10.
Calamia KT, Schirmer M, Melikoglu M. Major vessel involvement in Behcet disease. Curr Opin Rheumatol. 2005;17:1–8.
Uzun O, Akpolat T, Erkan L. Pulmonary vasculitis Behcet disease: a cumulative analysis. Chest. 2005;127:2243–53.
Seyahi E, Melikoglu M, Akman C, Hamuryudan V, Ozer H, Hatemi G, et al. Pulmonary artery involvement and associated lung disease in Behcet disease: a series of 47 patients. Medicine. 2012;91:35–48.
Gravallese EM, Corson JM, Coblyn JS, Pinkus GS, Weinblatt ME. Rheumatoid aortitis: a rarely recognized but clinically significant entity. Medicine. 1989;68:95–106.
Kawasuji M, Hetzer R, Oelert H, Stauch G, Borst HG. Aortic valve replacement and ascending aorta replacement in ankylosing spondylitis: report of three surgical cases and review of the literature. Thorac Cardiovasc Surg. 1982;30:310–4.
Stamp L, Lambie N, O'Donnell J. HLA-B27 associated spondyloarthropathy and severe ascending aortitis. J Rheumatol. 2000;27:2038–40.
Haynes BF, Kaiser-Kupfer MI, Mason P, Fauci AS. Cogan syndrome: studies in thirteen patients, long-term follow-up, and a review of the literature. Medicine. 1980;59:426–41.
Selim AG, Fulford LG, Mohiaddin RH, Sheppard MN. Active aortitis in relapsing polychondritis. J Clin Pathol. 2001;54:890–2.
Rho YH, Choi SJ, Choi YS, Lee YH, Ji JD, Song GG. Relapsing polychondritis with aortitis without valvular involvement. J Rheumatol. 2005;32:954–6.
Stone JR. Aortitis, periaortitis, and retroperitoneal fibrosis, as manifestations of IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:88–94. A comprehensive review on IgG4-related systemic disease as a cause of thoracic aortitis, inflammatory abdominal aortic aneurysm or peri-aortitis, and retroperitoneal fibrosis.
Compliance with Ethics Guidelines
Conflict of Interest
Soumya Chatterjee and Carmela D. Tan declare that they have no conflict of interest. Scott D. Flamm has been a consultant for Bayer Healthcare on the Cardiac Imaging Advisory Board. E. Rene Rodriguez declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Peripheral Vascular Disease
Rights and permissions
About this article

Cite this article
Chatterjee, S., Flamm, S.D., Tan, C.D. et al. Clinical Diagnosis and Management of Large Vessel Vasculitis: Takayasu Arteritis. Curr Cardiol Rep 16, 499 (2014). https://doi.org/10.1007/s11886-014-0499-y
Published:
DOI: https://doi.org/10.1007/s11886-014-0499-y
Keywords
- Large vessel vasculitis
- Takayasu arteritis
- Claudication
- Stenosis
- Occlusion
- Ectasia
- Aneurysm
- Dissection
- Acute phase reactant
- ESR
- CRP
- Interleukin-6
- Computerized tomographic angiogram (CTA)
- Magnetic resonance angiogram (MRA)
- Fluorodeoxyglucose(18F)-positron emission tomography (FDG-PET)
- Vascular Doppler ultrasound
- Glucocorticoid
- Prednisone
- Methylprednisolone
- Methotrexate
- Azathioprine
- Leflunomide
- Cyclophosphamide
- Tocilizumab
- Rituximab
- TNF inhibitors
- Angioplasty
- Stent
- Bypass