
Abstract
Recent studies involving HDL-raising therapeutics have greatly changed our understanding of this field. Despite effectively raising HDL-C levels, niacin remains of uncertain clinical benefit. Synthetic niacin receptor agonists are unlikely to raise HDL-C or have other beneficial effects on plasma lipids. Despite the failure in phase 3 of 2 CETP inhibitors, 2 potent CETP inhibitors that raise HDL-C levels by >100 % (and reduce LDL-C substantially) are in late stage clinical development. Infusions of recombinant HDL containing ‘wild-type’ apoA-I or apoA-I Milano, as well as autologous delipidated HDL, all demonstrated promising early results, and remain in clinical development. A small molecule that causes upregulation of endogenous apoA-I production is also in clinical development. Finally, upregulation of macrophage cholesterol efflux pathways through agonism of liver X receptors or antagonism of miR-33 remains of substantial interest. The field of HDL therapeutics is poised to transition from the ‘HDL-cholesterol hypothesis’ to the ‘HDL flux hypothesis’ in which the impact on flux from macrophage to feces is deemed to be of greater therapeutic benefit than the increase in steady-state concentrations of HDL cholesterol.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite aggressive LDL-reducing therapy, there remains substantial residual cardiovascular risk [1, 2]. The strong inverse association of plasma levels of high density lipoprotein (HDL) cholesterol with coronary artery disease (CAD) led to the development of the “HDL cholesterol hypothesis” that intervention to raise HDL cholesterol will result in reduced risk of CAD. The most popular mechanistic theory underlying the HDL cholesterol hypothesis has been the concept of “reverse cholesterol transport”; that HDL promotes cholesterol efflux from arterial macrophage foam cells and transports it to the liver for biliary excretion [3]. Recent discoveries have provided new insights into the complex metabolic and anti-atherosclerotic pathways of HDL. However, recent studies – including 2 randomized, placebo-controlled intervention trials [4••, 5] and a large genetic association analysis [6••] – call for a careful re-examination of the HDL cholesterol hypothesis. Here we review the current status of HDL-targeted therapies in the context of a re-evaluation of the HDL cholesterol hypothesis.
Niacin and Niacin Receptor Agonists
Nicotinic acid, or niacin, is the most effective HDL-raising drug currently on the market. In addition to raising HDL-C levels, niacin reduces triglycerides (TG), LDL-C, and lipoprotein(a) [Lp(a)]. New published clinical data involving niacin and its effects on lipids over the last 2 years has primarily involved studies of combination therapy as well as with co-administration with laropiprant, an antagonist of the prostaglandin receptor that mediates niacin-induced flushing. In a multiple-arm study in hyperlipidemic patients, the addition of extended-release niacin (ER-niacin) to the combination of ezetimibe/simvastatin significantly improved multiple lipid parameters compared with ezetimibe/simvastatin alone [7]. Metabolic syndrome patients randomized to ER-niacin in combination with laropiprant lowered TG and LDL-C and increased HDL-C significantly compared with placebo [8]. The combination of niacin/laropiprant plus a statin compared with doubling of the statin dose showed that combination treatment was associated with a significantly greater decrease in TG and LDL-C and a significantly greater increase in HDL-C [9]. An analysis of 4 trials of niacin/laropiprant revealed that this combination led to significantly larger decreases in TG, LDL-C, non-HDL-C, apoB, and Lp(a), and significantly greater increases in HDL-C and apoA-I compared with placebo or active comparator [10].
There has been considerable work on the molecular mechanisms of niacin action, both regarding its lipid and atherosclerosis effects as well as its cutaneous side effects, over the last several years. The discovery of the niacin receptor GPR109A created substantial excitement with regard to a better understanding of niacin’s molecular mechanisms of action. Activation of GPR109A on skin cells such as Langerhans cells and keratinocytes promotes synthesis of prostaglandin D2 (PGD2) and prostaglandin E2 (PGE2), which subsequently induce cutaneous capillary vasodilation by binding to DP1 and EP2/4 receptors [11–13]. Co-administration of the DP1 antagonist laropiprant significantly reduces but does not eliminate niacin-induced skin symptoms [14]. It is also clear that activation of GPR109A on adipocytes mediates suppression of lipolysis and release of free fatty acids [15].
Nevertheless, recent evidence strongly indicates that activation of GPR109A does not mediate niacin’s effects on plasma lipid and lipoprotein concentrations. Studies in a partially “humanized” mouse model showed that niacin reduces TG and LDL-C levels even when the GPR109A receptor is genetically deleted [16••]. Even more compelling are data in humans using synthetic agonists of GPR109A. Administration of several synthetic GPR109A agonists in humans effectively suppressed lipolysis and plasma free fatty acids but had minimal to no effect on TG, LDL-C, or HDL-C levels [16••, 17]. These results suggest that niacin modulates plasma lipids through mechanism(s) independent of its receptor GPR109A, and have brought into serious question the wisdom of developing synthetic GPR109A agonists as a therapeutic strategy. Interestingly, however, activation of GPR109A by niacin mediates certain anti-inflammatory effects such as macrophage recruitment into atherosclerotic plaques and the peritoneum. Provocatively, administration of niacin to atherosclerosis-prone mice decreased atherosclerotic plaques despite having minimal effects on cholesterol levels; this effect was dependent on expression of GPR109A in hematopoietic cells [18]. These results suggest that niacin may inhibit atherogenesis through activation of its receptor in macrophages or other hematopoietic cells and independently of effects on plasma lipid levels.
While niacin clearly improves all major lipid fractions, the major question has been whether it provides additional cardiovascular benefit when added to a statin, particularly in patients with low levels of HDL-C, and CAD. A randomized clinical trial in the pre-statin era in men with hypercholesterolemia and CAD indicated clinical benefit with reduction in cardiovascular event rates [19, 20]. Studies using vascular imaging measures suggested a benefit of combined simvastatin plus niacin on angiographic coronary disease and on carotid intimal medial thickness [21, 22]. Indeed, in 2010 2 published meta-analyses of randomized controlled trials of niacin concluded that niacin therapy was associated with significant reduction in major coronary events, stroke, and overall cardiovascular events, and led to the regression of coronary atherosclerosis and carotid intima thickness [23, 24].
Two trials were launched several years ago to test the incremental benefit of niacin added to a statin in patients with CAD and low HDL-C on cardiovascular events. The Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) trial randomized 3414 statin-treated CAD patients with low HDL-C to niacin vs placebo. The trial was halted early for futility: while niacin modestly but significantly increased HDL-C levels, there was no difference between the 2 groups in terms of CV events [4••]. The Heart Protection Study 2 - Treatment of High density lipoprotein to Reduce the Incidence of Vascular Events (HPS2-THRIVE) trial is a much larger trial with an arguably more appropriate comparative effectiveness design [25], and is scheduled to report by the end of 2012 [26, 27]. Thus, at the current time the clinical benefit of niacin, particularly with regard to the benefit of its HDL-raising properties, is uncertain.
Cholesteryl Ester Transfer Protein (CETP) Inhibitors
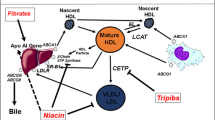
The cholesteryl ester transfer protein (CETP) exchanges cholesteryl esters primarily from HDL for triglycerides primarily from VLDL [28]. Genetic CETP deficiency causes marked elevation in HDL-C levels, leading to the concept that pharmacologic inhibition of CETP would raise HDL-C levels and, according to the “HDL cholesterol hypothesis,” reduce cardiovascular risk. The first CETP inhibitor to be tested in humans, torcetrapib, did substantially increase HDL-C levels [29]. However, in a large clinical outcome trial, subjects randomized to torcetrapib had increased cardiovascular events and total mortality compared with those allocated to placebo [30]. This outcome has been widely attributed to off-target effects of torcetrapib, including increased aldosterone production and raising of blood pressure [31, 32].
The next CETP inhibitor to enter a phase 3 clinical outcome trial was dalcetrapib. Less potent than torcetrapib, it increased HDL-C by an average of about 25 %–30 % in clinical trials [33–35]. The dal-PLAQUE trial tested the effect of dalcetrapib on atherosclerotic plaques using arterial positron-emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging (MRI). It focused largely on ruling out an adverse torcetrapib-like effect and found no adverse effects on vessel wall/arterial inflammation -- but little evidence of beneficial effects [36]. The dal-VESSEL trial tested the effect of dalcetrapib on vascular function and found no effect, adverse or beneficial, on NO-dependent endothelial function, inflammation, or oxidative stress [37]. Finally, the dal-OUTCOMES trial was a phase 3 outcomes trial testing the effect of dalcetrapib on cardiovascular events. It was terminated early due to futility; no details have yet been reported. This result would appear to challenge the simplistic HDL cholesterol hypothesis that raising HDL-C will reduce cardiovascular risk, and also poses additional questions about CETP inhibition as a therapeutic strategy.
However, at least 2 CETP inhibitors, both more potent than dalcetrapib, remain in clinical development. Anacetrapib inhibits CETP by forming a tight reversible bond [38]. At a dose of 150 mg daily it not only raises HDL-C by > 100 % but also reduces LDL-C by about 40 % and Lp(a) by up to 50 % [39, 40]. In a phase 2b trial, 589 dyslipidemic patients were administered anacetrapib, atorvastatin, or placebo in various combinations with anacetrapib causing substantial increases in HDL-C and apoA-I and reductions in LDL-C and apoB [41•]. In a detailed study of lipoprotein subfractions, anacetrapib increased large HDL particles enriched with CE, apoA-I and apoC-III [42]. There are also some data suggesting that anacetrapib may improve HDL function and reverse cholesterol transport. HDL from subjects who received 300 mg anacetrapib daily for 8 weeks promoted greater cholesterol efflux from foam cells than HDL from subjects given placebo [43]. A detailed study in hamsters showed that anacetrapib promoted macrophage-specific reverse cholesterol transport (RCT) compared with placebo [44].
The Determining the Efficacy and Tolerability of CETP Inhibition with Anacetrapib (DEFINE) trial randomized 1623 patients with CAD or at high-risk for CAD who had achieved LDL-C treatment goals with statin therapy to 100 mg of anacetrapib or placebo [45, 46•]. Anacetrapib treatment resulted in a 138 % increase in HDL-C, a 40 % reduction in LDL-C, and a 36 % decrease in Lp(a) [46•]. After 76 weeks of follow-up, no increases in blood pressure, serum aldosterone levels, or cardiovascular events were observed. The Randomized Evaluation of the Effects of Anacetrapib Through Lipid-modification (REVEAL) trial is now enrolling and will randomize 30,000 subjects with CAD, cerebrovascular atherosclerotic disease, or peripheral artery disease to anacetrapib 100 mg, or placebo to formally assess the impact on cardiovascular events. The estimated study completion date is January 2017.
Evacetrapib is another potent CETP inhibitor in clinical development. Administration of evacetrapib in a 12-week randomized trial of 398 dyslipidemic patients in doses of 30–500 mg daily as monotherapy increased HDL-C from 54 %–129 % and decreased LDL-C from 14 %–36 %. Addition of evacetrapib 100 mg daily to statin therapy produced similar HDL-C increases and yielded further LDL-C reductions [47•]. A large phase III clinical outcomes trial is apparently planned to determine the effect of evacetrapib in reducing cardiovascular events [48].
In summary, the first CETP inhibitor (torcetrapib) to enter phase 3 increased CV events and mortality due most likely to off-target effects. The second CETP inhibitor (dalcetrapib) to enter phase 3 failed due to lack of efficacy in reducing cardiovascular risk, with details still unknown. There is a theoretical case to be made that CETP inhibition may not be an optimal mechanism to target HDL. However, at least 2 potent CETP inhibitors (anacetrapib and evacetrapib) are still in clinical development. In addition to raising HDL-C levels considerably more than dalcetrapib, they also substantially reduce LDL-C and Lp(a) levels. Thus, even if the HDL-C raising is of marginal benefit, these potent CETP inhibitors may reduce CV risk due to their effects on LDL-C and Lp(a).
Infusions of apoA-I-Containing Recombinant HDL Particles
ApoA-I is the most abundant protein in HDL. Lipid-poor apoA-I is effective at promoting cholesterol efflux from macrophages by serving as the preferred “acceptor” of cholesterol from the adenosine triphosphate binding cassette transporter 1 (ABCA1) transporter. Animal studies are strongly supportive of the concept that overexpression or injection of apoA-I can reduce or even regress atherosclerotic plaque. Thus there is interest in the concept of infusing apoA-I-containing recombinant HDL particles in humans. Small clinical studies using coronary imaging support the concept of intravenous apoA-I infusion for reducing cardiovascular risk [49–51, 52•].
One approach uses apoA-I purified from human plasma and complexed with phosphatidylcholine derived from soybean, a preparation often termed “recombinant HDL” (rHDL) [51, 53]. A randomized, placebo-controlled study involved the administration in 145 patients with acute coronary syndrome of 4 weekly infusions of this type of rHDL (termed CSL-111). Serial intravascular ultrasound (IVUS) was used to assess the impact on coronary atherosclerosis. Infusion of rHDL was found to reduce atheroma volume by 3.4 % compared with baseline, although this was not significantly different than placebo [51]. Another study in patients with lower extremity peripheral artery disease utilized a single CSL-111 infusion followed by percutaneous superficial femoral artery revascularization 5-7 days following CSL-111 infusion. There were significant reductions in lipid content and endothelial adhesion molecule expression in plaque excised by atherectomy [54]. Among patients with diabetes, infusion of CSL-111 increased HDL-C up to 40 %, inhibited ex vivo platelet aggregation, and reduced monocyte activation and neutrophil adhesion [55, 56]. A reformulated version of CSL-111, called CSL-112, has been reported in preclinical studies to provide greater cholesterol efflux capacity as well as reduced hepatotoxicity compared with CSL-111, and is currently in clinical development [57].
ApoA-I Milano is a naturally-occurring mutation in apoA-I that has a cysteine to arginine substitution at amino acid 173. It is associated with very low levels of HDL-C, but despite this is not associated with increased atherosclerotic disease, giving rise to the concept that this mutant apoA-I may actually be more anti-atherogenic [58, 59]. Recombinant apoA-I Milano complexed with phospholipid has therefore also been studied for its effects on atherosclerosis in animal models and in humans. In mice and rabbits it has been shown to reduce atherosclerosis [60–63], though not to a greater extent than wild-type rHDL [64–67]. In a human study in subjects with CAD, 5 weekly doses of apoA-I Milano rHDL or placebo were administered and coronary atheroma was investigated by IVUS at the beginning and end of the study. Total atheroma volume was significantly reduced compared with baseline but no significant difference was observed with placebo [49]. ApoA-I Milano-containing rHDL infusion is still being investigated.
Another approach to apoA-I infusion utilizes autologous delipidated HDL [52•]. A device was invented that involves collection of plasma by apheresis over 1.5–2 hours followed by selective removal of lipids from HDL using organic solvents. The delipidated HDL is subsequently reinfused over 1 hour. In a non-human primate study this approach resulted in a significant reduction in aortic atheroma volume by IVUS [68]. In a clinical study of 28 patients with acute coronary syndrome, 7 weekly infusions of autologous delipidated HDL decreased total atheroma volume significantly from baseline though not from the control group [52•].
Upregulation of Endogenous apoA-I Production
Upregulation of endogenous apoA-I is conceptually highly attractive. RVX-208 is a synthetic small molecule that increases the transcription of the apoA-I gene. In a monkey model, administration of the compound RVX-208 over approximately 2 months significantly increased plasma apoA-I levels up to 60 % in a dose-dependent manner [69]. A small human study showed a significant increase in plasma apoA-I levels of 10 %, as well as augmentation of cholesterol efflux capacity [69]. In a Phase 2 trial of RVX-208, 299 statin-treated CAD patients were randomized to placebo or 3 different treatment doses for 12 weeks. While there was an increase in apoA-I levels compared with baseline, there was no statistically significant change in apoA-I compared with placebo [70•]. An ongoing phase 2b trial of RVX-208 involves 172 statin-treated patients randomized to placebo or RVX-208 100 mg twice daily for 24 weeks [71]. In another phase 2b trial, the effect of RVX-208 on coronary atherosclerosis is being assessed by IVUS [72]. It will be of substantial interest to determine whether this approach has beneficial effects on coronary disease.
Enhancing Macrophage Cholesterol Efflux Through Upregulation of ABC Transporters
Liver X receptors (LXRs) are nuclear receptors that act as cholesterol sensors and regulate expression of genes involved in cholesterol metabolism. Activation of LXRs by natural and synthetic agonists has been demonstrated to promote mobilization of intracellular cholesterol, increase macrophage cholesterol efflux via macrophage ABCA1 and ABCG1, and augment intestinal HDL generation [73–75]. Dyslipidemic hamsters treated with the LXR agonist GW3965 had increased macrophage-to-feces RCT, but also increased TG and LDL-C [76]. In a rabbit model of atherosclerosis, combination therapy with the LXR agonist LXR-623, and simvastatin induced plaque regression, while simvastatin alone, and LXR monotherapy at low and medium doses attenuated plaque progression [77].
Therapeutic development of LXR agonists has been hindered by hepatic steatosis and increased plasma triglyceride concentrations reported in preclinical studies of these drugs [78]. Dissociating LXR efficacy and toxicity might be possible owing to the differential effects of LXR agonism by receptor isoform and by tissue-specific effects. Administration of a nonselective LXR agonist to LXRα-deficient mice stimulated macrophage ABCA1 expression and cholesterol efflux without inducing fatty liver and with minimal upregulation of hepatic triglyceride synthesis [79]. Liver-specific LXRα knockout mice have increased atherosclerosis and decreased RCT. Interestingly, synthetic LXR agonist rescue in these mice still led to anti-atherogenic activity despite the lack of hepatic LXRalpha; it also did not increase plasma TG but still increased plasma HDL [80]. The LXR agonist AZ876 decreased atherosclerotic lesion size in APOE*3 Leiden mice at low dose without increasing TG or causing liver steatosis; at high dose, lesion size was decreased to an even greater degree and also there were decreased cytokine levels/vessel wall inflammatory markers, but with the addition of increased plasma TG [81].
A second approach to LXR agonist development might be to selectively activate intestinal LXR. An intestine-specific LXRα/β agonist, GW6340, promoted macrophage-specific reverse cholesterol transport, augmenting the fecal excretion of radiolabeled sterol by 52 % via increased intestinal HDL production and intestinal excretion of HDL-derived cholesterol [82].
Another mechanism for increasing ABCA1 and ABCG1 expression is through the microRNA miR-33. MicroRNAs are short non-coding sequences of RNA that inhibit gene expression by binding to complementary 3’ untranslated regions of mRNAs and causing translational repression and/or mRNA destabilization [83]. MiR-33 is encoded within an intron of the gene encoding the sterol regulatory element binding transcription factor 2 (SREBF2) and suppresses macrophage and hepatocyte expression of ABCA1 and ABCG1, thus reducing circulating HDL-C levels and macrophage efflux to apoA-I [84]. Silencing of miR-33 with an antisense oligonucleotide (ASO) was associated with greater macrophage and hepatocyte expression of ABCA1 and increased HDL-C levels. In a mouse model of atherosclerosis, administration of an ASO to miR-33 significantly increased HDL-C, promoted macrophage-specific reverse cholesterol transport, and induced atheroma regression [85••]. In a non-human primate model of dyslipidemia, subcutaneous delivery of anti-miR-33 ASO over a 12-week period increased HDL-C up to 50 % [86•]. Greater macrophage cholesterol efflux was observed following incubation of foam cells with serum obtained from treated monkeys compared with equivalent volumes of serum isolated from control monkeys, correlating with the HDL-C levels in the 2 groups. Thus, anti-miR-33 therapy is another potential approach to promoting macrophage cholesterol efflux and RCT.
Conclusions
Recent events have brought into question the simple “HDL cholesterol hypothesis” that raising HDL cholesterol levels will reduce cardiovascular risk. The HPS2-THRIVE trial with niacin and the trials with CETP inhibitors anacetrapib and evacetrapib will provide the next wave of critical clinical data of HDL-directed strategies in large contemporary cohorts managed with aggressive medical therapy. It may be time to modify the “HDL cholesterol hypothesis” to the “HDL flux hypothesis”: Intervention to promote cholesterol efflux and reverse cholesterol transport will reduce CAD risk. Clinical outcomes studies of interventions that promote cholesterol efflux and reverse cholesterol transport are ultimately required to test this hypothesis.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Boden WE, O'Rourke RA, Teo KK, et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med. 2007;356:1503–16.
Frye RL, August P, Brooks MM, et al. A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med. 2009;360:2503–15.
deGoma EM, deGoma RL, Rader DJ. Beyond high-density lipoprotein cholesterol levels evaluating high-density lipoprotein function as influenced by novel therapeutic approaches. J Am Coll Cardiol. 2008;51:2199–211.
•• Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67. AIM-HIGH trial stopped early due to futility: niacin increased HDL-C but did not decrease CV events.
Roche provides update on Phase III study of dalcetrapib. Roche. 2012. http://www.roche.com/media/media_releases/med-cor-2012-05-07.htm. Accessed 11 Sept 2012.
•• Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80. A large human genetics study showing that genetic variants that affect HDL-C levels do not necessarily associate with CAD risk. The most striking example was a variant in the gene LIPG encoding the enzyme endothelial lipase that raises HDL-C but was not associated with protection from CAD.
Fazio S, Guyton JR, Polis AB, et al. Long-term safety and efficacy of triple combination ezetimibe/simvastatin plus extended-release niacin in patients with hyperlipidemia. Am J Cardiol. 2010;105:487–94.
Bays HE, Shah A, Lin J, McCrary Sisk C, Paolini JF, Maccubbin D. Efficacy and tolerability of extended-release niacin/laropiprant in dyslipidemic patients with metabolic syndrome. J Clin Lipidol. 2010;4(6):515–21.
Shah S, Ceska R, Gil-Extremera B, et al. Efficacy and safety of extended-release niacin/laropiprant plus statin vs doubling the dose of statin in patients with primary hypercholesterolaemia or mixed dyslipidaemia. Int J Clin Pract. 2010;64:727–38.
Bays HE, Shah A, Lin J, Sisk CM, Dong Q, Maccubbin D. Consistency of extended-release niacin/laropiprant effects on Lp(a), ApoB, non-HDL-C, Apo A1, and ApoB/ApoA1 ratio across patient subgroups. Am J Cardiovasc Drugs. 2012;12:197–206.
Dunbar RL, Gelfand JM. Seeing red: flushing out instigators of niacin-associated skin toxicity. J Clin Invest. 2010;120:2651–5.
Hanson J, Gille A, Zwykiel S, et al. Nicotinic acid- and monomethyl fumarate-induced flushing involves GPR109A expressed by keratinocytes and COX-2-dependent prostanoid formation in mice. J Clin Invest. 2010;120:2910–9.
Digby JE, Ruparelia N, Choudhury RP. Niacin in cardiovascular disease: recent preclinical and clinical developments. Arterioscler Thromb Vasc Biol. 2012;32:582–8
Maccubbin D, Koren MJ, Davidson M, et al. Flushing profile of extended-release niacin/laropiprant vs gradually titrated niacin extended-release in patients with dyslipidemia with and without ischemic cardiovascular disease. Am J Cardiol. 2009;104:74–81.
Tunaru S, Kero J, Schaub A, et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nature Medicine. 2003;9:352–5.
•• Lauring B, Taggart AK, Tata JR, et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Science Translational Med, 2012;4(148). A comprehensive series of studies in mice and humans that convincingly shows that the niacin receptor GPR109A is not responsible for the favorable lipid changes associated with niacin therapy.
Lai E, Waters G, Tata J, et al. Effects of a niacin receptor partial agonist, MK-0354, on plasma free fatty acids, lipids, and cutaneous flushing in humans. J Clin Lipidol. 2008;2:375–83.
Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest. 2011;121(3):1163–73
Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345:1583–92.
Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8:1245–55.
Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial biology for the investigation of the treatment effects of reducing cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110:3512–7.
Villines TC, Stanek EJ, Devine PJ, et al. The ARBITER 6-HALTS Trial (Arterial biology for the investigation of the treatment effects of reducing cholesterol 6-HDL and LDL treatment strategies in atherosclerosis): final results and the impact of medication adherence, dose, and treatment duration. J Am Coll Cardiol. 2010;55:2721–6.
Duggal JK, Singh M, Attri N, et al. Effect of niacin therapy on cardiovascular outcomes in patients with coronary artery disease. J Cardiovasc Pharmacol Ther. 2010;15:158–66.
Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis. 2010;210:353–61.
Brinton EA. Search and rescue for hypotheses surviving AIM-HIGH, the niacin therapy earthquake: still problematic after the primary publication. J Clin Lipidol. 2012;6:312–7.
National Heart, Lung, and Blood Institute; Abbott. Niacin plus statin to prevent vascular events. In: ClinicalTrials.gov. National Library of Medicine. 2011. http://www.clinicaltrials.gov/ct2/show/NCT00120289. Accessed 11 Sept 2012.
University of Oxford; Merck. Treatment of HDL to reduce the incidence of vascular events HPS2-THRIVE. In: ClinicalTrials.gov. National Library of Medicine. 2010. http://www.clinicaltrials.gov/ct2/show/NCT00461630. Accessed 11 Sept 2012.
Neeli H, Rader DJ. Cholesteryl ester transfer protein (CETP) inhibitors: is there life after torcetrapib? Cardiol Clin. 2008;26:537–46.
Brousseau ME, Schaefer EJ, Wolfe ML, et al. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N Engl J Med. 2004;350:1505–15.
Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22.
Rader DJ. Illuminating HDL–is it still a viable therapeutic target? N Engl J Med. 2007;357:2180–3.
Barter P. Lessons learned from the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial. Am J Cardiol. 2009;104(10 Suppl):10E–5E.
de Grooth GJ, Kuivenhoven JA, Stalenhoef AF, et al. Efficacy and safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in humans: a randomized phase II dose-response study. Circulation. 2002;105(18):2159–65.
Kuivenhoven JA, de Grooth GJ, Kawamura H, et al. Effectiveness of inhibition of cholesteryl ester transfer protein by JTT-705 in combination with pravastatin in type II dyslipidemia. Am J Cardiol. 2005;95(9):1085–8.
Stein EA, Roth EM, Rhyne JM, Burgess T, Kallend D, Robinson JG. Safety and tolerability of dalcetrapib (RO4607381/JTT-705): results from a 48-week trial. Eur Heart J. 2010;31:480–8.
Fayad ZA, Mani V, Woodward M, et al. Safety and efficacy of dalcetrapib on atherosclerotic disease using novel non-invasive multimodality imaging (dal-PLAQUE): a randomized clinical trial. Lancet. 2011;378:1547–59.
Luscher TF, Taddei S, Kaski JC, et al. Vascular effects and safety of dalcetrapib in patients with or at risk of coronary heart disease: the dal-VESSEL randomized clinical trial. Eur Heart J. 2012;33:857–65.
Ranalletta M, Bierilo KK, Chen Y, et al. Biochemical characterization of cholesteryl ester transfer protein inhibitors. J Lipid Res. 2010;51:2739–52.
Krishna R, Anderson MS, Bergman AJ, et al. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: 2 double-blind, randomized placebo-controlled phase I studies. Lancet. 2007;370:1907–14.
Bloomfield D, Carlson GL, Sapre A, et al. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am Heart J. 2009;157(e352):352–60.
• Dansky HM, Bloomfield D, Gibbons P, et al. Efficacy and safety after cessation of treatment with the cholesteryl ester transfer protein inhibitor anacetrapib (MK-0859) in patients with primary hypercholesterolemia or mixed hyperlipidemia. Am Heart J. 2011;162:708–16. Phase 2b trial of anacetrapib showing increases in HDL-C and apoA-I as well as reduced LDL-C and apoB.
Krauss RM, Wojnooski K, Orr J, et al. Changes in lipoprotein subfraction concentration and composition in healthy individuals treated with the CETP inhibitor anacetrapib. J Lipid Res. 2012;53:540–7.
Yvan-Charvet L, Kling J, Pagler T, et al. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol. 2010;30:1430–8.
Castro-Perez J, Briand F, Gagen K, et al. Anacetrapib promotes reverse cholesterol transport and bulk cholesterol excretion in Syrian golden hamsters. J Lipid Res. 2011;52:1965–73.
Cannon CP, Dansky HM, Davidson M, et al. Design of the DEFINE trial: determining the EFficacy and tolerability of CETP INhibition with AnacEtrapib. Am Heart J. 2009;158(e513):513–19.
• Cannon CP, Shah S, Dansky HM, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Eng J Med. 2010;363:2406–15. DEFINE trial of anacetrapib showing increased HDL-C, decreased LDL-C, and decreased Lp(a) without concerning off-target effects or a "torcetrapib-like" effect on CV events or mortality.
• Nicholls SJ, Brewer HB, Kastelein JJ, et al. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA:J Am Med Assoc. 2011;306:2099–109. Evacetrapib trial showing substantially increased HDL-C and decreased LDL-C across a dose range of evacetrapib.
Nicholls SJ. Evacetrapib. Current Cardiol Reports. 2012;14:245–50.
Nissen SE, Tsunoda T, Tuzcu EM, et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–300.
Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004;291(9):1071–80.
Tardif JC, Gregoire J, L’Allier PL, et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297:1675–82.
• Waksman R, Torguson R, Kent KM, et al. A first-in-man, randomized, placebo-controlled study to evaluate the safety and feasibility of autologous delipidated high-density lipoprotein plasma infusions in patients with acute coronary syndrome. J Am Coll Cardiol. 2010;55:2727–35. Autologous delipidated HDL significantly decreased total atheroma volume in ACS patients vs baseline.
Lerch PG, Fortsch V, Hodler G, Bolli R. Production and characterization of a reconstituted high density lipoprotein for therapeutic applications. Vox Sang. 1996;71:155–64.
Shaw JA, Bobik A, Murphy A, et al. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103:1084–91.
Patel S, Drew BG, Nakhla S, et al. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J Am Coll Cardiol. 2009;53:962–71.
Calkin AC, Drew BG, Ono A, et al. Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation. 2009;120:2095–104.
CSL Limited. A single ascending dose study examining the safety and pharmacokinetic profile of reconstituted high density lipoprotein (CSL112) administered to patients. In: ClinicalTrials.gov. National Library of Medicine. 2012. http://www.clinicaltrials.gov/ct2/show/NCT01499420. Accessed 11 Sept 2012.
Gualandri V, Franceschini G, Sirtori CR, et al. A-I Milano apoprotein identification of the complete kindred and evidence of a dominant genetic transmission. Am J Hum Genet. 1985;37:1083–97.
Sirtori CR, Calabresi L, Franceschini G, et al. Cardiovascular status of carriers of the apolipoprotein A-I Milano mutant: the Limone sul Garda study. Circulation. 2001;103:1949–54.
Ameli S, Hultgardh-Nilsson A, Cercek B, et al. Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation. 1994;90:1935–41.
Shah PK, Nilsson J, Kaul S, et al. Effects of recombinant apolipoprotein A-I Milano on aortic atherosclerosis in apolipoprotein E-deficient mice. Circulation. 1998;97:780–5.
Shah PK, Yano J, Reyes O, et al. High-dose recombinant apolipoprotein A-I(milano) mobilizes tissue cholesterol and rapidly reduces plaque lipid and macrophage content in apolipoprotein e-deficient mice. Potential implications for acute plaque stabilization. Circulation. 2001;103:3047–50.
Parolini C, Marchesi M, Lorenzon P, et al. Dose-related effects of repeated ETC-216 (recombinant apolipoprotein A-I Milano/1-palmitoyl-2-oleoyl phosphatidylcholine complexes) administrations on rabbit lipid-rich soft plaques: in vivo assessment by intravascular ultrasound and magnetic resonance imaging. J Am Coll Cardiol. 2008;51:1098–103.
Weibel GL, Alexander ET, Joshi MR, et al. Wild-type ApoA-I and the Milano variant have similar abilities to stimulate cellular lipid mobilization and efflux. Arterioscler Thromb Vasc Biol. 2007;27:2022–9.
Lebherz C, Sanmiguel J, Wilson JM, Rader DJ. Gene transfer of wild-type apoA-I and apoA-I Milano reduce atherosclerosis to a similar extent. Cardiovasc Diabetol. 2007;6:15.
Parolini C, Chiesa G, Gong E, et al. Apolipoprotein A-I and the molecular variant apoA-I Milano: evaluation of the antiatherogenic effects in knock-in mouse model. Atherosclerosis. 2005;183:222–9.
Alexander ET, Weibel GL, Joshi MR, et al. Macrophage reverse cholesterol transport in mice expressing ApoA-I Milano. Arterioscler Thromb Vasc Biol. 2009;29:1496–501.
Sacks FM, Rudel LL, Conner A, et al. Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. J Lipid Res. 2009;50:894–907.
Bailey D, Jahagirdar R, Gordon A, et al. RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J Am Coll Cardiol. 2010;55:2580–9.
• Nicholls SJ, Gordon A, Johansson J, et al. Efficacy and safety of a novel oral inducer of apolipoprotein A-I synthesis in statin-treated patients with stable coronary artery disease a randomized controlled trial. J Am Coll Cardiol. 2011;57:1111–9. RVX-208 was shown to increase plasma apoA-I levels vs baseline.
Resverlogix Corp; The Cleveland Clinic. The study of quantitative serial trends in lipids with apolipoproteinA-I stimulation (SUSTAIN). In: ClinicalTrials.gov. National Library of Medicine. 2012. http://www.clinicaltrials.gov/ct2/show/NCT01423188. Accessed 11 Sept 2012.
Resverlogix Corp; The Cleveland Clinic. ApoA-I synthesis stimulation and intravascular ultrasound for coronary atheroma regression evaluation (ASSURE I). In: ClinicalTrials.gov. National Library of Medicine. 2012. http://www.clinicaltrials.gov/ct2/show/NCT01067820. Accessed 11 Sept 2012.
Rigamonti E, Helin L, Lestavel S, et al. Liver X receptor activation controls intracellular cholesterol trafficking and esterification in human macrophages. Circ Res. 2005;97:682–9.
Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem. 2000;275:28240–5.
Brunham LR, Kruit JK, Iqbal J, et al. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J Clin Invest. 2006;116:1052–62.
Briand F, Treguier M, Andre A, et al. Liver X receptor activation promotes macrophage-to-feces reverse cholesterol transport in a dyslipidemic hamster model. J Lipid Res. 2010;51:763–70.
Giannarelli C, Cimmino G, Connolly TM, et al. Synergistic effect of liver X receptor activation and simvastatin on plaque regression and stabilization: a magnetic resonance imaging study in a model of advanced atherosclerosis. Eur Heart J. 2012;33:264–73.
Grefhorst A, Elzinga BM, Voshol PJ, et al. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J Biol Chem. 2002;277:34182–90.
Lund EG, Peterson LB, Adams AD, et al. Different roles of liver X receptor alpha and beta in lipid metabolism: effects of an alpha-selective and a dual agonist in mice deficient in each subtype. Biochem Pharmacol. 2006;71:453–63.
Zhang Y, Breevoort SR, Angdisen J, et al. Liver LXRalpha expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J Clin Invest. 2012;122:1688–99.
van der Hoorn J, Linden D, Lindahl U, et al. Low dose of the liver X receptor agonist, AZ876, reduces atherosclerosis in APOE*3Leiden mice without affecting liver or plasma triglyceride levels. Br J Pharmacol. 2011;162:1553–63.
Yasuda T, Grillot D, Billheimer JT, et al. Tissue-specific liver X receptor activation promotes macrophage reverse cholesterol transport in vivo. Arterioscler Thromb Vasc Biol. 2010;30:781–6.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Rayner KJ, Suarez Y, Davalos A, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–3.
•• Rayner KJ, Sheedy FJ, Esau CC, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–31. Treatment with an ASO against miR-33 in mice was shown to increase macrophage reverse cholesterol transport and regress atherosclerosis.
• Rayner KJ, Esau CC, Hussain FN, et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. 2011;478:404–7. Treatment with an ASO against miR-33 in non-human primates was shown to increase HDL-C levels and reduce VLDL concentrations.
Acknowledgments
This paper was supported in part by the National Center for Research Resources, Grant TL1RR024133, and is now at the National Center for Advancing Translational Sciences, Grant TL1R000138. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosures
Conflicts of interest: D. B. Larach: none; E.M. deGoma: none; D.J. Rader: serves as a consultant to several companies that are developing therapies targeting HDL, including Amgen, AstraZeneca, Bristol-Myers-Squibb, CSL, Eli Lilly, Johnson & Johnson, Merck & Co, Regulus, and Novartis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Larach, D.B., deGoma, E.M. & Rader, D.J. Targeting High Density Lipoproteins in the Prevention of Cardiovascular Disease?. Curr Cardiol Rep 14, 684–691 (2012). https://doi.org/10.1007/s11886-012-0317-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-012-0317-3